Joint analysis of paired and unpaired multiomic data with MultiVI#
MultiVI is used for the joint analysis of scRNA and scATAC-seq datasets that were jointly profiled (multiomic / paired) and single-modality datasets (only scRNA or only scATAC). MultiVI uses the paired data as an anchor to align and merge the latent spaces learned from each individual modality.
This tutorial walks through how to read multiomic data, create a joint object with paired and unpaired data, set-up and train a MultiVI model, visualize the resulting latent space, and run differential analyses.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
[notice] A new release of pip is available: 24.2 -> 24.3.1
[notice] To update, run: pip install --upgrade pip
import gzip
import os
import tempfile
from pathlib import Path
import matplotlib.pyplot as plt
import mudata as md
import muon
import numpy as np
import pooch
import scanpy as sc
import scvi
import seaborn as sns
import torch
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.2.0
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Data acquisition#
First we download a sample multiome dataset from 10X. We’ll use this throughout this tutorial. Importantly, MultiVI assumes that there are shared features between the datasets. This is trivial for gene expression datasets, which generally use the same set of genes as features. For ATAC-seq peaks, this is less trivial, and often requires preprocessing steps with other tools to get all datasets to use a shared set of peaks. That can be achieved with tools like SnapATAC, ArchR, and CellRanger in the case of 10X data.
Important
MultiVI requires the datasets to use shared features. scATAC-seq datasets need to be processed to use a shared set of peaks.
Next, we’ll read the data into an Mudata object, which is a container for multiple AnnData objects.
Reading the data into an MuData object can be done with the muon read_10x_h5 function:
Note
We could have also used Anndata object. However, this is less recommended starting v1.2.1 and we aim that the use of anndata with MultiVI will be deprecated starting scvi-tools v1.4.0. To see MultiVI Anndata tutorial, see RTD previous versions.
# read multiomic data
url = "https://cf.10xgenomics.com/samples/cell-arc/2.0.0/10k_PBMC_Multiome_nextgem_Chromium_X/10k_PBMC_Multiome_nextgem_Chromium_X_filtered_feature_bc_matrix.h5"
mdata = muon.read_10x_h5("data/multiome10k.h5mu", backup_url=url)
Added `interval` annotation for features from data/multiome10k.h5mu
#We add batch artificially for this tutorial purpose
mdata["rna"].obs["batch"] = ['batch_0'] * int(mdata.n_obs/2) + ['batch_1'] * (mdata.n_obs - int(mdata.n_obs/2))
#We also add size factor to be used during minification later in this tutorial
mdata.obs["size_factor_rna"] = mdata["rna"].X.sum(1)
mdata.obs["size_factor_atac"] = (mdata["atac"].X.sum(1) + 1) / (np.max(mdata["atac"].X.sum(1)) + 1.01)
mdata
MuData object with n_obs × n_vars = 10970 × 148344
obs: 'size_factor_rna', 'size_factor_atac'
var: 'gene_ids', 'feature_types', 'genome', 'interval'
2 modalities
rna: 10970 x 36601
obs: 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'interval'
atac: 10970 x 111743
var: 'gene_ids', 'feature_types', 'genome', 'interval'We can see that there are 2 modalities in this multiome data, rna and atac. For more information on this dataset: sample multiome data from 10X of 10K PBMCs.
Data Processing#
We can use scanpy functions to handle, filter, and manipulate the data. In our case, we might want to filter out peaks & genes that are rarely detected, to make the model train faster. In this case we keep the top 4000 for each modality and filter out non-chromosomal regions. We store those in a new modality for reproducibility.
sc.pp.highly_variable_genes(
mdata.mod["rna"],
n_top_genes=4000,
flavor="seurat_v3",
)
mdata.mod["rna_subset"] = mdata.mod["rna"][:, mdata.mod["rna"].var["highly_variable"]].copy()
# Filter out non-chromosomal regions
mask = mdata.mod["rna_subset"].var["interval"].str.startswith("chr")
mdata.mod["rna_subset"] = mdata.mod["rna_subset"][:, mask].copy()
sc.pp.highly_variable_genes(
mdata.mod["atac"],
n_top_genes=4000,
flavor="seurat_v3",
)
mdata.mod["atac_subset"] = mdata.mod["atac"][:, mdata.mod["atac"].var["highly_variable"]].copy()
# Filter out non-chromosomal regions
mask = mdata.mod["atac_subset"].var["interval"].str.startswith("chr")
mdata.mod["atac_subset"] = mdata.mod["atac_subset"][:, mask].copy()
mdata.update()
mdata
MuData object with n_obs × n_vars = 10970 × 156340
obs: 'size_factor_rna', 'size_factor_atac'
var: 'gene_ids', 'feature_types', 'genome', 'interval'
4 modalities
rna: 10970 x 36601
obs: 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
atac: 10970 x 111743
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
rna_subset: 10970 x 4001
obs: 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
atac_subset: 10970 x 3995
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'mdata.mod["rna_subset"].var
| gene_ids | feature_types | genome | interval | highly_variable | highly_variable_rank | means | variances | variances_norm | |
|---|---|---|---|---|---|---|---|---|---|
| HES4 | ENSG00000188290 | Gene Expression | GRCh38 | chr1:999980-1000172 | True | 210.0 | 0.063446 | 0.173748 | 2.174311 |
| ISG15 | ENSG00000187608 | Gene Expression | GRCh38 | chr1:1001137-1013497 | True | 848.0 | 0.204102 | 0.426294 | 1.466590 |
| TNFRSF18 | ENSG00000186891 | Gene Expression | GRCh38 | chr1:1205679-1206592 | True | 1859.0 | 0.036554 | 0.056736 | 1.263666 |
| TNFRSF4 | ENSG00000186827 | Gene Expression | GRCh38 | chr1:1214152-1214153 | True | 2126.0 | 0.053965 | 0.082601 | 1.226946 |
| AL162741.1 | ENSG00000260179 | Gene Expression | GRCh38 | chr1:1251333-1251334 | True | 3940.0 | 0.001003 | 0.001184 | 1.105230 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| LINC00278 | ENSG00000231535 | Gene Expression | GRCh38 | chrY:3002886-3003001 | True | 3015.0 | 0.390246 | 0.750511 | 1.153820 |
| AC010737.1 | ENSG00000229308 | Gene Expression | GRCh38 | chrY:4036496-4036497 | True | 921.0 | 0.000365 | 0.000547 | 1.446148 |
| NLGN4Y | ENSG00000165246 | Gene Expression | GRCh38 | chrY:14522751-14524574 | True | 1655.0 | 0.019508 | 0.030616 | 1.289717 |
| TTTY14 | ENSG00000176728 | Gene Expression | GRCh38 | chrY:19076023-19077395 | True | 255.0 | 0.146855 | 0.406821 | 2.044043 |
| TTTY10 | ENSG00000229236 | Gene Expression | GRCh38 | chrY:20575221-20575362 | True | 2629.0 | 0.035278 | 0.051176 | 1.182238 |
4001 rows × 9 columns
mdata.mod["atac_subset"].var
| gene_ids | feature_types | genome | interval | highly_variable | highly_variable_rank | means | variances | variances_norm | |
|---|---|---|---|---|---|---|---|---|---|
| chr1:633578-634591 | chr1:633578-634591 | Peaks | GRCh38 | chr1:633578-634591 | True | 1603.0 | 1.308751 | 4.189740 | 1.232472 |
| chr1:1098910-1099873 | chr1:1098910-1099873 | Peaks | GRCh38 | chr1:1098910-1099873 | True | 799.0 | 0.296809 | 0.935326 | 1.287371 |
| chr1:1201046-1201913 | chr1:1201046-1201913 | Peaks | GRCh38 | chr1:1201046-1201913 | True | 3971.0 | 0.891796 | 2.632014 | 1.169175 |
| chr1:1216749-1217579 | chr1:1216749-1217579 | Peaks | GRCh38 | chr1:1216749-1217579 | True | 1471.0 | 0.219690 | 0.656080 | 1.238325 |
| chr1:1629814-1630730 | chr1:1629814-1630730 | Peaks | GRCh38 | chr1:1629814-1630730 | True | 2596.0 | 1.805925 | 5.822209 | 1.197963 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| chrY:56727693-56728598 | chrY:56727693-56728598 | Peaks | GRCh38 | chrY:56727693-56728598 | True | 2097.0 | 0.002370 | 0.005464 | 1.211180 |
| chrY:56734351-56735237 | chrY:56734351-56735237 | Peaks | GRCh38 | chrY:56734351-56735237 | True | 42.0 | 0.004467 | 0.015022 | 1.694357 |
| chrY:56763067-56763960 | chrY:56763067-56763960 | Peaks | GRCh38 | chrY:56763067-56763960 | True | 53.0 | 0.002461 | 0.007743 | 1.648426 |
| chrY:56836418-56837345 | chrY:56836418-56837345 | Peaks | GRCh38 | chrY:56836418-56837345 | True | 254.0 | 0.026800 | 0.080237 | 1.382211 |
| chrY:56857542-56858287 | chrY:56857542-56858287 | Peaks | GRCh38 | chrY:56857542-56858287 | True | 1378.0 | 0.005014 | 0.012465 | 1.243371 |
3995 rows × 9 columns
mdata.mod["atac_subset"].var.interval.nunique()
3995
Setup and Training MultiVI#
We can now set up and train the MultiVI model!
Now we run setup_mudata, which is the MuData analog to setup_anndata. The caveat of this workflow is that we need to provide this function which modality of the mdata object contains each piece of data.
So for example, if the batch information is in mdata.mod["rna"].obs["batch"], in the modalities argument we specify that the batch_key can be found in the "rna_subset" modality of the MuData object (see example in the TotalVI tutorial).
In our case, the main batch annotation, by mudata definition, correspond to the modality of the cells.
Important
MultiVI requires the main batch annotation to correspond to the modality of the samples. Other batch annotation, such as in the case of multiple RNA-only batches, can be specified using categorical_covariate_keys.
scvi.model.MULTIVI.setup_mudata(
mdata,
batch_key="batch", # the batch is here: mdata.mod["rna_subset"].obs["batch"]
size_factor_key = ["size_factor_rna", "size_factor_atac"],
modalities={
"rna_layer": "rna_subset",
"atac_layer": "atac_subset",
"batch_key": "rna_subset",
}
)
Note
Specify the modality of each argument via the modalities dictionary, which maps layer/key arguments to MuData modalities.
Prepare and run the model
model = scvi.model.MULTIVI(mdata)
model.view_anndata_setup()
Anndata setup with scvi-tools version 1.2.0.
Setup via `MULTIVI.setup_anndata` with arguments:
{ │ 'rna_layer': None, │ 'atac_layer': None, │ 'protein_layer': None, │ 'batch_key': 'batch', │ 'size_factor_key': ['size_factor_rna', 'size_factor_atac'], │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None, │ 'idx_layer': None, │ 'modalities': {'rna_layer': 'rna_subset', 'atac_layer': 'atac_subset', 'batch_key': 'rna_subset'} }
Summary Statistics ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━┓ ┃ Summary Stat Key ┃ Value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━┩ │ n_atac │ 3995 │ │ n_batch │ 2 │ │ n_cells │ 10970 │ │ n_extra_categorical_covs │ 0 │ │ n_extra_continuous_covs │ 0 │ │ n_labels │ 1 │ │ n_size_factor │ 2 │ │ n_vars │ 4001 │ └──────────────────────────┴───────┘
Data Registry ┏━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Registry Key ┃ scvi-tools Location ┃ ┡━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ X │ adata.mod['rna_subset'].X │ │ atac │ adata.mod['atac_subset'].X │ │ batch │ adata.mod['rna_subset'].obs['_scvi_batch'] │ │ ind_x │ adata.obs['_indices'] │ │ labels │ adata.obs['_scvi_labels'] │ │ size_factor │ adata.obsm['_scvi_size_factor'] │ └──────────────┴────────────────────────────────────────────┘
batch State Registry ┏━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['batch'] │ batch_0 │ 0 │ │ │ batch_1 │ 1 │ └────────────────────┴────────────┴─────────────────────┘
labels State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['_scvi_labels'] │ 0 │ 0 │ └───────────────────────────┴────────────┴─────────────────────┘
size_factor State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['size_factor_rna'] │ │ adata.obs['size_factor_atac'] │ └───────────────────────────────┘
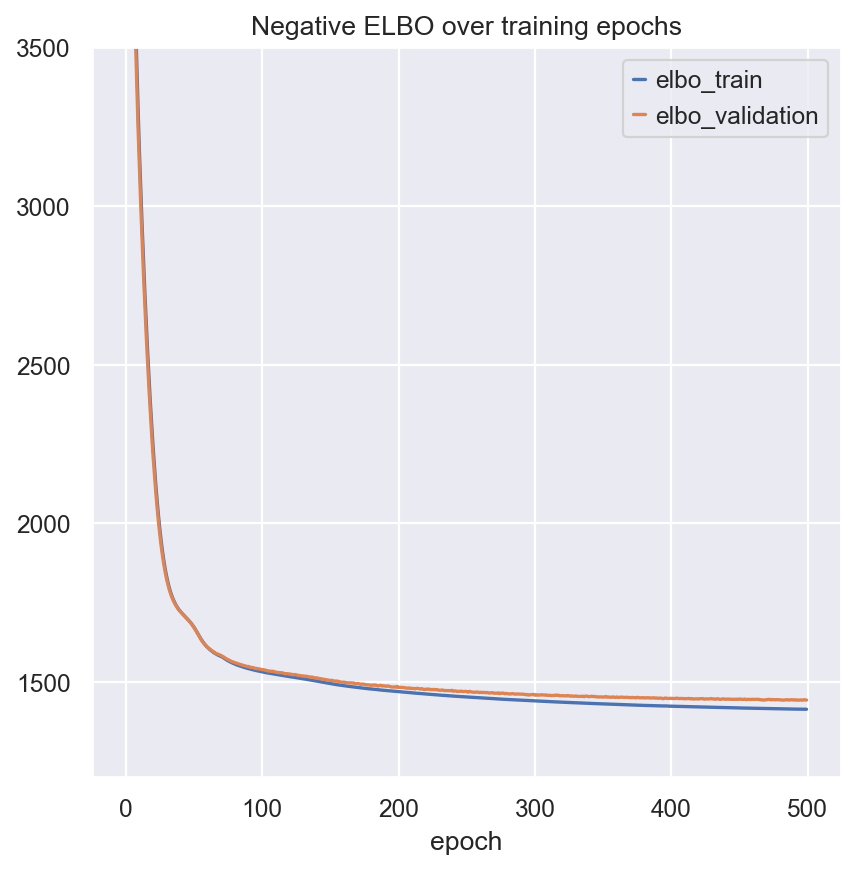
model.train()
fig, ax = plt.subplots(1, 1)
model.history["elbo_train"].plot(ax=ax, label="train")
model.history["elbo_validation"].plot(ax=ax, label="validation")
ax.set(title="Negative ELBO over training epochs", ylim=(1200, 3500))
ax.legend()
<matplotlib.legend.Legend at 0x323a9dbd0>
Save and Load MultiVI models#
Saving and loading models is similar to all other scvi-tools models, and is very straight forward:
model_dir = os.path.join(save_dir.name, "multivi_pbmc10k")
model.save(model_dir, overwrite=True, save_anndata=True)
model.view_setup_args(os.path.join(save_dir.name, "multivi_pbmc10k"))
INFO File /var/folders/l9/pf9bmk9x5nx429m28xmk34740000gq/T/tmpckri987f/multivi_pbmc10k/model.pt already
downloaded
Setup via `MULTIVI.setup_anndata` with arguments:
{ │ 'rna_layer': None, │ 'atac_layer': None, │ 'protein_layer': None, │ 'batch_key': 'batch', │ 'size_factor_key': ['size_factor_rna', 'size_factor_atac'], │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None, │ 'idx_layer': None, │ 'modalities': {'rna_layer': 'rna_subset', 'atac_layer': 'atac_subset', 'batch_key': 'rna_subset'} }
model_loaded = scvi.model.MULTIVI.load(model_dir, adata=mdata)
INFO File /var/folders/l9/pf9bmk9x5nx429m28xmk34740000gq/T/tmpckri987f/multivi_pbmc10k/model.pt already
downloaded
Extracting and visualizing the latent space#
We can now use the get_latent_representation to get the latent space from the trained model, and visualize it using scanpy functions:
MULTIVI_LATENT_KEY = "X_multivi"
MULTIVI_CLUSTERS_KEY = "leiden_mutliVI"
mdata.obsm[MULTIVI_LATENT_KEY] = model.get_latent_representation()
mdata.update()
sc.pp.neighbors(mdata, use_rep=MULTIVI_LATENT_KEY)
sc.tl.umap(mdata, min_dist=0.2)
sc.tl.leiden(mdata, key_added=MULTIVI_CLUSTERS_KEY)
mdata.update()
sc.pl.umap(mdata, color=["rna_subset:batch","leiden_mutliVI"])
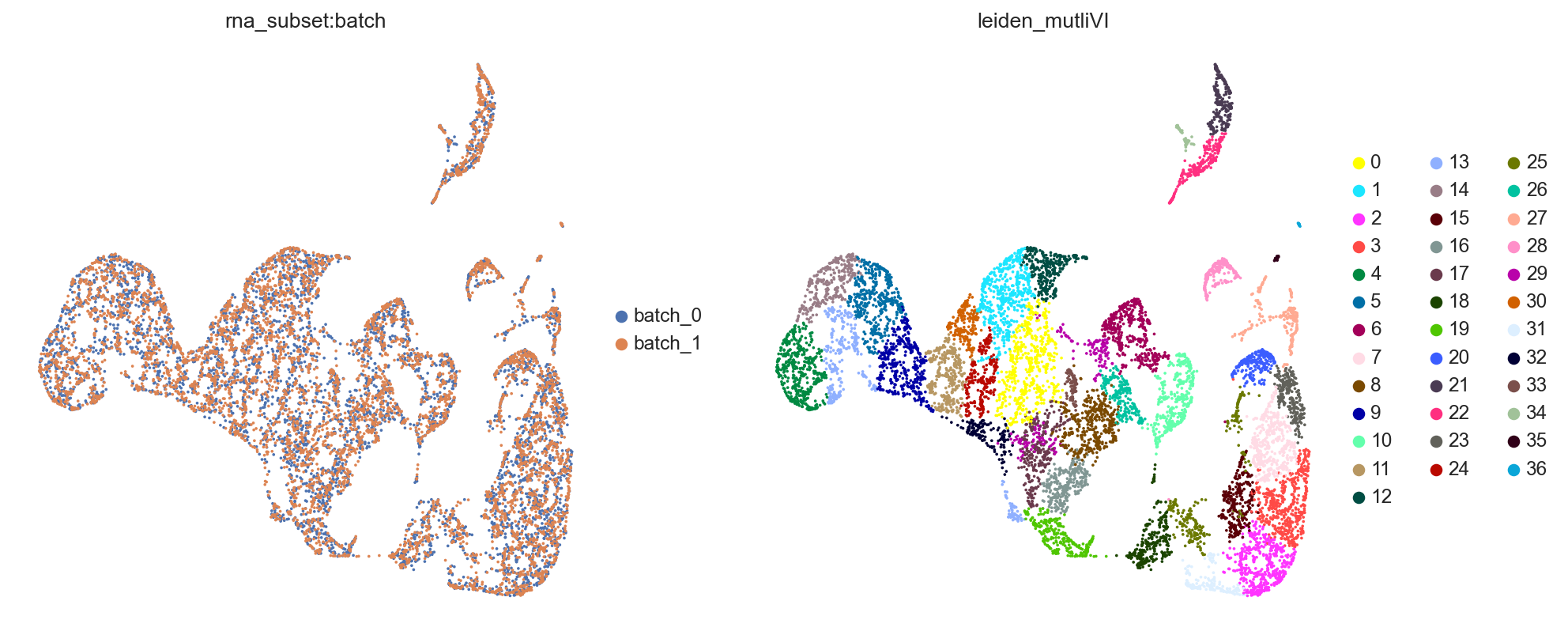
muon has another plotting functions which can pull data from either modality of the MuData object.
Impute missing modalities#
In a well-mixed space, MultiVI can seamlessly impute the missing modalities for single-modality cells.
First, imputing expression and accessibility is done with get_normalized_expression and get_accessibility_estimates, respectively.
We’ll demonstrate this by imputing gene expression for all cells in the dataset (including those that are ATAC-only cells):
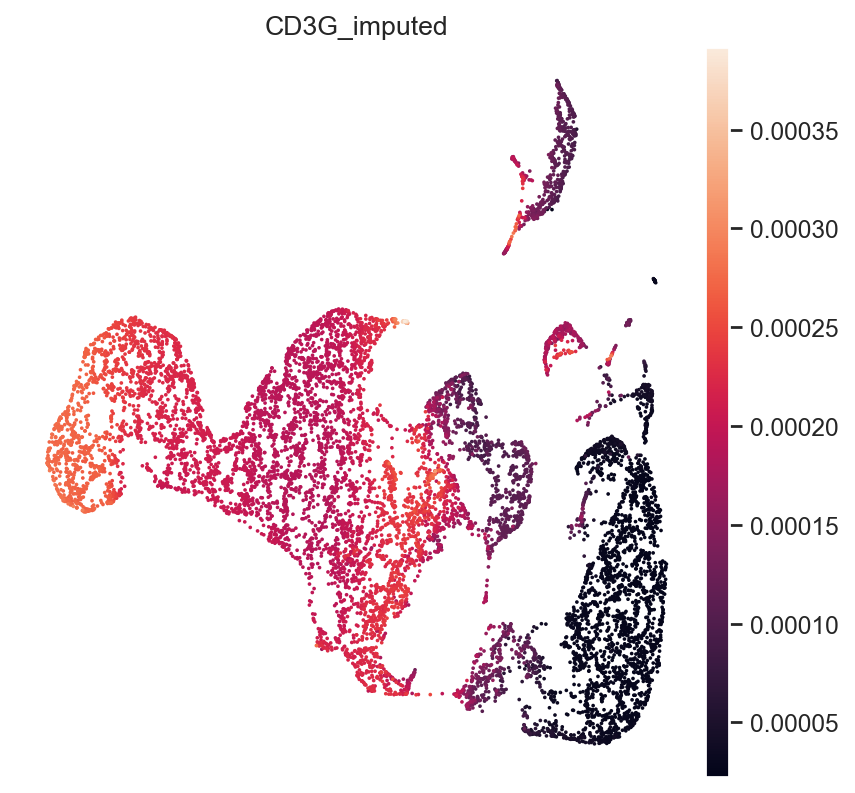
imputed_expression = model.get_normalized_expression()
We can demonstrate this on some known marker genes:
First, T-cell marker CD3.
gene_idx = np.where(mdata["rna_subset"].var.index == "CD3G")[0]
mdata.obs["CD3G_imputed"] = imputed_expression.iloc[:, gene_idx]
sc.pl.umap(mdata, color="CD3G_imputed")
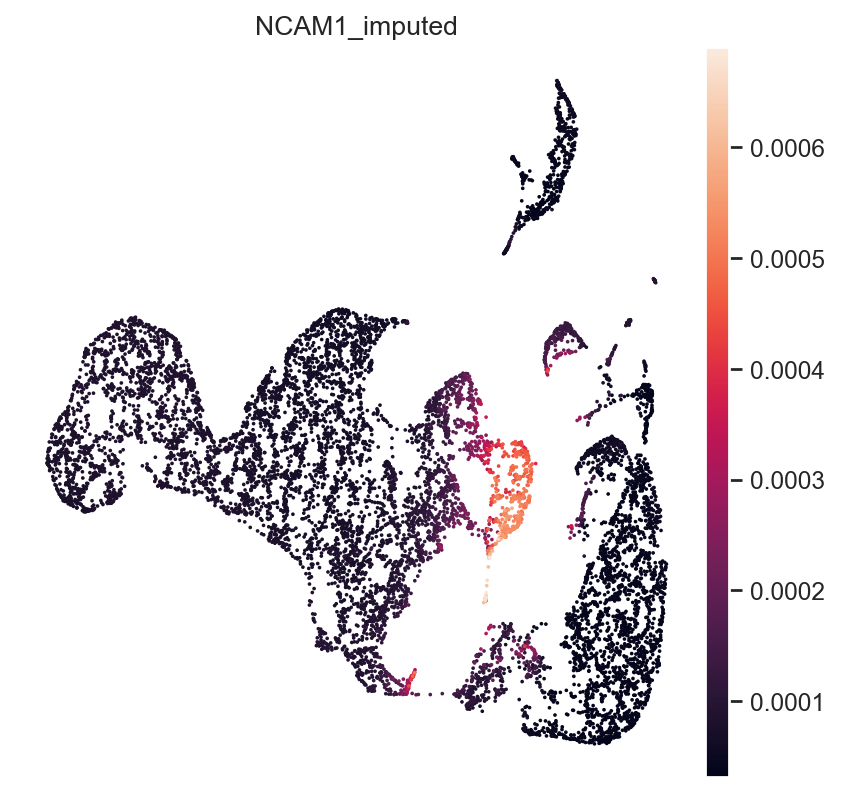
Next, NK-Cell marker gene NCAM1 (CD56):
gene_idx = np.where(mdata["rna_subset"].var.index == "NCAM1")[0]
mdata.obs["NCAM1_imputed"] = imputed_expression.iloc[:, gene_idx]
sc.pl.umap(mdata, color="NCAM1_imputed")
Finally, B-Cell Marker CD19:
gene_idx = np.where(mdata["rna_subset"].var.index == "CD19")[0]
mdata.obs["CD19_imputed"] = imputed_expression.iloc[:, gene_idx]
sc.pl.umap(mdata, color="CD19_imputed")
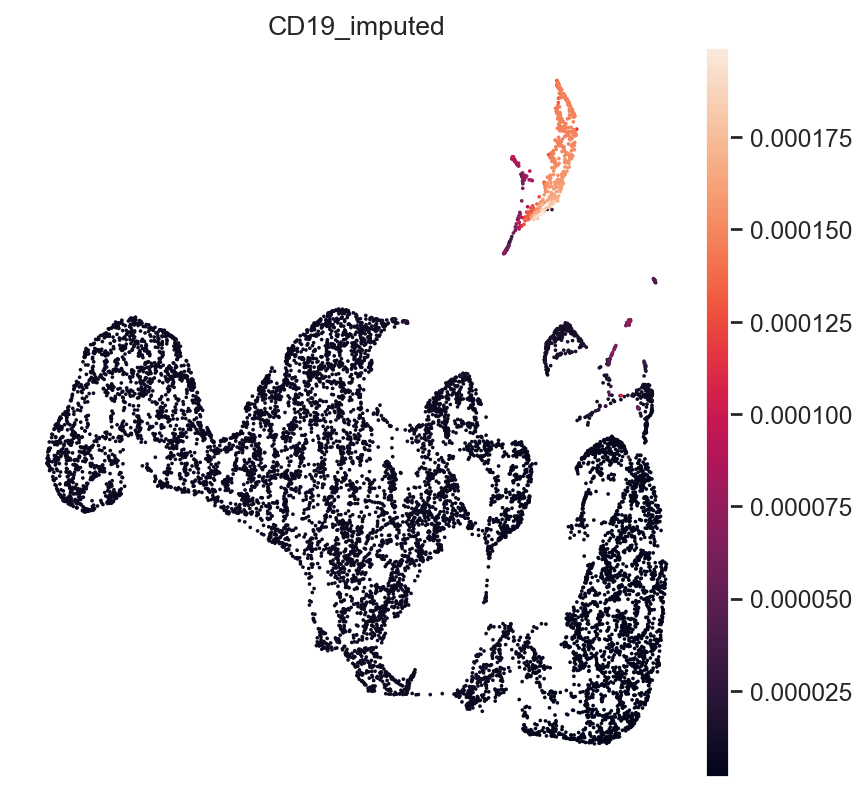
All three marker genes clearly identify their respective populations. Importantly, the imputed gene expression profiles are stable and consistent within that population, even though many of those cells only measured the ATAC profile of those cells.
Minification of MultiVI models#
We will show here how to apply minification to the mudata of the trained multivi model. The reader is first encouraged to read the minification tutorial, which can be found here: (https://docs.scvi-tools.org/en/stable/tutorials/notebooks/hub/minification..html)
model
MultiVI Model with the following params: n_genes: 4001, n_regions: 3995, n_proteins: 0, n_hidden: 63, n_latent: 7, n_layers_encoder: 2, n_layers_decoder: 2, dropout_rate: 0.1, latent_distribution: normal, deep injection: False, gene_likelihood: zinb, gene_dispersion:gene, Mod.Weights: equal, Mod.Penalty: Jeffreys, protein_dispersion: protein Training status: Trained Model's adata is minified?: False Model's adata is minified?: False
Let’s also save a reference to model.adata. We’ll see later that this remains unchanged because minification is not an inplace procedure.
bdata = model.adata
Note that, as expected, “Model’s adata is minified” is False.
To minify the data, all we need to do is:
get the latent representation and store it in the mudata
call
model.minify_mudata()
qzm, qzv = model.get_latent_representation(give_mean=False, return_dist=True)
model.adata.obsm["X_latent_qzm"] = qzm
model.adata.obsm["X_latent_qzv"] = qzv
model.minify_mudata()
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
INFO Generating sequential column names
INFO Generating sequential column names
model
MultiVI Model with the following params: n_genes: 4001, n_regions: 3995, n_proteins: 0, n_hidden: 63, n_latent: 7, n_layers_encoder: 2, n_layers_decoder: 2, dropout_rate: 0.1, latent_distribution: normal, deep injection: False, gene_likelihood: zinb, gene_dispersion:gene, Mod.Weights: equal, Mod.Penalty: Jeffreys, protein_dispersion: protein Training status: Trained Model's adata is minified?: True Model's adata is minified?: True
As expected, “Model’s adata is minified” is now True. Also, we can check the model’s minified_data_type:
model.minified_data_type
'latent_posterior_parameters'
model.adata
MuData object with n_obs × n_vars = 10970 × 156340
obs: 'size_factor_rna', 'size_factor_atac', '_indices', '_scvi_labels', 'leiden_mutliVI', 'CD3G_imputed', 'NCAM1_imputed', 'CD19_imputed', '_multivi_observed_lib_size'
var: 'gene_ids', 'feature_types', 'genome', 'interval'
uns: '_scvi_manager_uuid', 'neighbors', 'umap', 'leiden_mutliVI', 'leiden_mutliVI_colors', '_scvi_adata_minify_type', '_scvi_uuid'
obsm: '_scvi_size_factor', 'X_multivi', 'X_umap', 'X_latent_qzm', 'X_latent_qzv', '_multivi_latent_qzm', '_multivi_latent_qzv'
obsp: 'distances', 'connectivities'
4 modalities
rna: 10970 x 36601
obs: 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
atac: 10970 x 111743
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
rna_subset: 10970 x 4001
obs: 'batch', '_scvi_batch'
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'
atac_subset: 10970 x 3995
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'hvg'First, let’s check that the original mudata was not modified (minification is not inplace):
model.adata is bdata
False
scvi.data._utils._is_minified(model.adata)
True
Lets verify the different modalities are empty now
model.adata.mod["rna"].X
<10970x36601 sparse matrix of type '<class 'numpy.float64'>'
with 0 stored elements in Compressed Sparse Row format>
Save and load minified model#
Just like a regular model, you can save the model and its minified data, and load them back in.
We also verify the actual size on disk
minified_model_path = os.path.join(save_dir.name, "multivi_minified")
model.save(minified_model_path, save_anndata=True, overwrite=True)
before = os.path.getsize(os.path.join(model_dir, "mdata.h5mu")) // (1024 * 1024)
after = os.path.getsize(os.path.join(minified_model_path, "mdata.h5mu")) // (1024 * 1024)
print(f"AnnData size before minification: {before} MB")
print(f"AnnData size after minification: {after} MB")
AnnData size before minification: 970 MB
AnnData size after minification: 68 MB
# load saved model with saved (minified) adata
loaded_minify_model = scvi.model.MULTIVI.load(minified_model_path)
loaded_minify_model
INFO File /var/folders/l9/pf9bmk9x5nx429m28xmk34740000gq/T/tmpckri987f/multivi_minified/model.pt already
downloaded
MultiVI Model with the following params: n_genes: 4001, n_regions: 3995, n_proteins: 0, n_hidden: 63, n_latent: 7, n_layers_encoder: 2, n_layers_decoder: 2, dropout_rate: 0.1, latent_distribution: normal, deep injection: False, gene_likelihood: zinb, gene_dispersion:gene, Mod.Weights: equal, Mod.Penalty: Jeffreys, protein_dispersion: protein Training status: Trained Model's adata is minified?: True Model's adata is minified?: True
loaded_minify_model.adata["rna"].X
<10970x36601 sparse matrix of type '<class 'numpy.float64'>'
with 0 stored elements in Compressed Sparse Row format>
Which is a minified version.
Next, let’s load the model with a non-minified data.
loaded_model = scvi.model.MULTIVI.load(model_dir, adata=bdata)
loaded_model
INFO File /var/folders/l9/pf9bmk9x5nx429m28xmk34740000gq/T/tmpckri987f/multivi_pbmc10k/model.pt already
downloaded
MultiVI Model with the following params: n_genes: 4001, n_regions: 3995, n_proteins: 0, n_hidden: 63, n_latent: 7, n_layers_encoder: 2, n_layers_decoder: 2, dropout_rate: 0.1, latent_distribution: normal, deep injection: False, gene_likelihood: zinb, gene_dispersion:gene, Mod.Weights: equal, Mod.Penalty: Jeffreys, protein_dispersion: protein Training status: Trained Model's adata is minified?: False Model's adata is minified?: False
And this is not minified as expected.