Disentangled representation learning with DRVI#
DRVI (Moinfar & Theis, 2024) is an unsupervised deep generative model that learns an interpretable, disentangled latent representation of single-cell omics data. Disentanglement is induced in the decoder: the latent is split into distinct groups that are decoded separately and aggregated, so each factor tends to capture a distinct, biologically meaningful axis of variation.
In this notebook we analyze an immune dataset of 9 batches from four human peripheral blood and bone marrow studies with 16 annotated cell types, training DRVI with 128 latent dimensions, to showcase:
How to train DRVI
How to inspect the latent space (vanished dimensions, UMAP, heatmap)
How to run the interpretability pipeline and link latent dimensions to genes
The model in this tutorial is imported entirely from scvi-tools (scvi.external.DRVI).
But the visualization helpers (drvi.utils.pl.*) come from the companion drvi-py package, installed with pip install drvi-py.
Note
Running the following cell is necessary if running in Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
[notice] A new release of pip is available: 24.3.1 -> 26.1.2
[notice] To update, run: pip install --upgrade pip
To install drvi-py package
!pip install --quiet drvi-py
Imports#
import os
import tempfile
import warnings
warnings.filterwarnings("ignore")
import anndata as ad
# Helper functions from drvi-py package
import drvi
import scanpy as sc
import scvi
from matplotlib import pyplot as plt
from scvi.external import DRVI
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
print("Last run with drvi version:", drvi.__version__)
Last run with scvi-tools version: 1.4.3
Last run with drvi version: 0.2.6
sc.set_figure_params(figsize=(4, 4), frameon=False)
save_dir = tempfile.TemporaryDirectory()
plt.rcParams["figure.dpi"] = 100
%config InlineBackend.print_figure_kwargs={'facecolor': 'w'}
%config InlineBackend.figure_format='retina'
Download and load data#
We use the immune dataset (Luecken et al.) hosted on SCVERSE S3. The Villani batch is already removed because it contains non-count values, and 2000 batch-aware highly variable genes are selected.
adata_path = os.path.join(save_dir.name, "immune_hvg.h5ad")
adata = sc.read(
adata_path, backup_url="https://exampledata.scverse.org/scvi-tools/Immune_HVG_human.h5ad"
)
adata
AnnData object with n_obs × n_vars = 32484 × 2000
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue'
uns: 'batch_colors', 'final_annotation_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts'
obsp: 'connectivities', 'distances'
Train DRVI#
We register the count layer with scvi.external.DRVI.setup_anndata and train a model with 128 latent dimensions and two hidden layers of width 128 in both the encoder and the decoder. By default DRVI splits every latent dimension (n_split_latent = n_latent) with split_map splitting and logsumexp aggregation — the fully disentangled setting.
DRVI’s general hyperparameters are fully compatible with scVI (encoder width/depth, likelihood, covariate handling, and training arguments carry over directly). DRVI expects count data and uses a negative-binomial likelihood by default. Provide additional covariates via categorical_covariate_keys / continuous_covariate_keys in setup_anndata if needed.
Depending on the data and modality, users can also choose other appropriate likelihoods such as Normal (needs library- and log-normalized data) or Poisson.
Please refer to the API documentation for hyperparameter choices.
DRVI.setup_anndata(
adata,
layer="counts",
batch_key="batch",
)
model = DRVI(
adata,
n_latent=128,
n_hidden=128,
n_layers=2,
# depending on the variability of gene dispersions use 'gene' (default) or 'gene-batch'
# dispersion='gene-batch',
)
model
INFO The model has been initialized
Model with the following params: n_latent: 128 n_split_latent: 128 split_method: 'split_map' split_aggregation: 'logsumexp'. gene_likelihood: 'pnb' Training status: Not Trained Model's adata is minified?: False
n_epochs = 400
model.train(
max_epochs=n_epochs,
plan_kwargs={"n_epochs_kl_warmup": n_epochs},
)
Epoch 400/400: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 400/400 [09:07<00:00, 1.37s/it, v_num=1, train_loss=745]
Epoch 400/400: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 400/400 [09:07<00:00, 1.37s/it, v_num=1, train_loss=745]
model_path = os.path.join(save_dir.name, "drvi_model")
model.save(model_path, overwrite=True)
# model = DRVI.load(model_path, adata)
Latent space and interpretability scores#
We store the latent representation in a new AnnData object (embed), where each factor is a latent dimension. DRVI.set_latent_dimension_stats annotates embed.var with per-dimension statistics (reconstruction effect, vanished status, ordering, min/max), and DRVI.calculate_interpretability_scores stores per-gene scores in embed.varm. The scores are calculated as follows:
OOD (out-of-distribution): traverses each latent dimension through the decoder.
IND (in-distribution): averages each dimension’s effect over the observed cells.
Note:
The order of latent dimensions (DR 1, DR 2, …) is different from the order of columns in embed. Use embed.var['title'].
embed = ad.AnnData(model.get_latent_representation(), obs=adata.obs.copy())
model.set_latent_dimension_stats(embed, vanished_threshold=0.5)
model.calculate_interpretability_scores(embed, "OOD")
model.calculate_interpretability_scores(embed, "IND")
sc.pp.neighbors(embed, n_neighbors=10, use_rep="X", n_pcs=embed.X.shape[1])
sc.tl.umap(embed, spread=1.0, min_dist=0.5, random_state=123)
embed
AnnData object with n_obs × n_vars = 32484 × 128
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue', '_scvi_batch', '_scvi_labels'
var: 'original_dim_id', 'reconstruction_effect', 'order', 'max_value', 'mean', 'min', 'max', 'std', 'std_abs', 'title', 'vanished', 'vanished_positive_direction', 'vanished_negative_direction'
uns: 'neighbors', 'umap'
obsm: 'X_umap'
varm: 'OOD_min_possible_positive', 'OOD_min_possible_negative', 'OOD_max_possible_positive', 'OOD_max_possible_negative', 'OOD_combined_positive', 'OOD_combined_negative', 'IND_max_positive', 'IND_max_negative', 'IND_linear_weighted_mean_positive', 'IND_linear_weighted_mean_negative', 'IND_exp_weighted_mean_positive', 'IND_exp_weighted_mean_negative'
obsp: 'distances', 'connectivities'
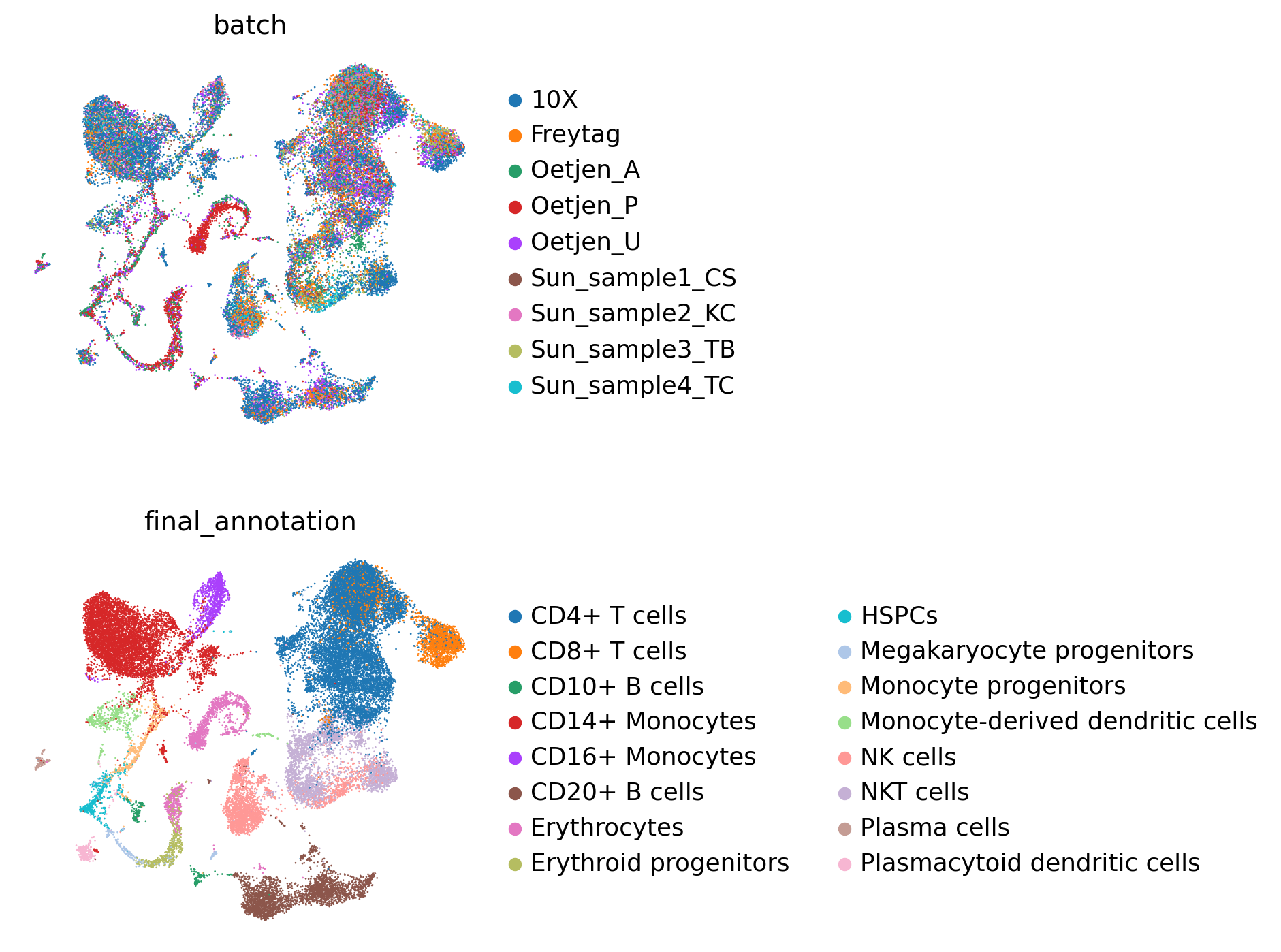
sc.pl.umap(embed, color=["batch", "final_annotation"], ncols=1, frameon=False)
Latent dimension stats#
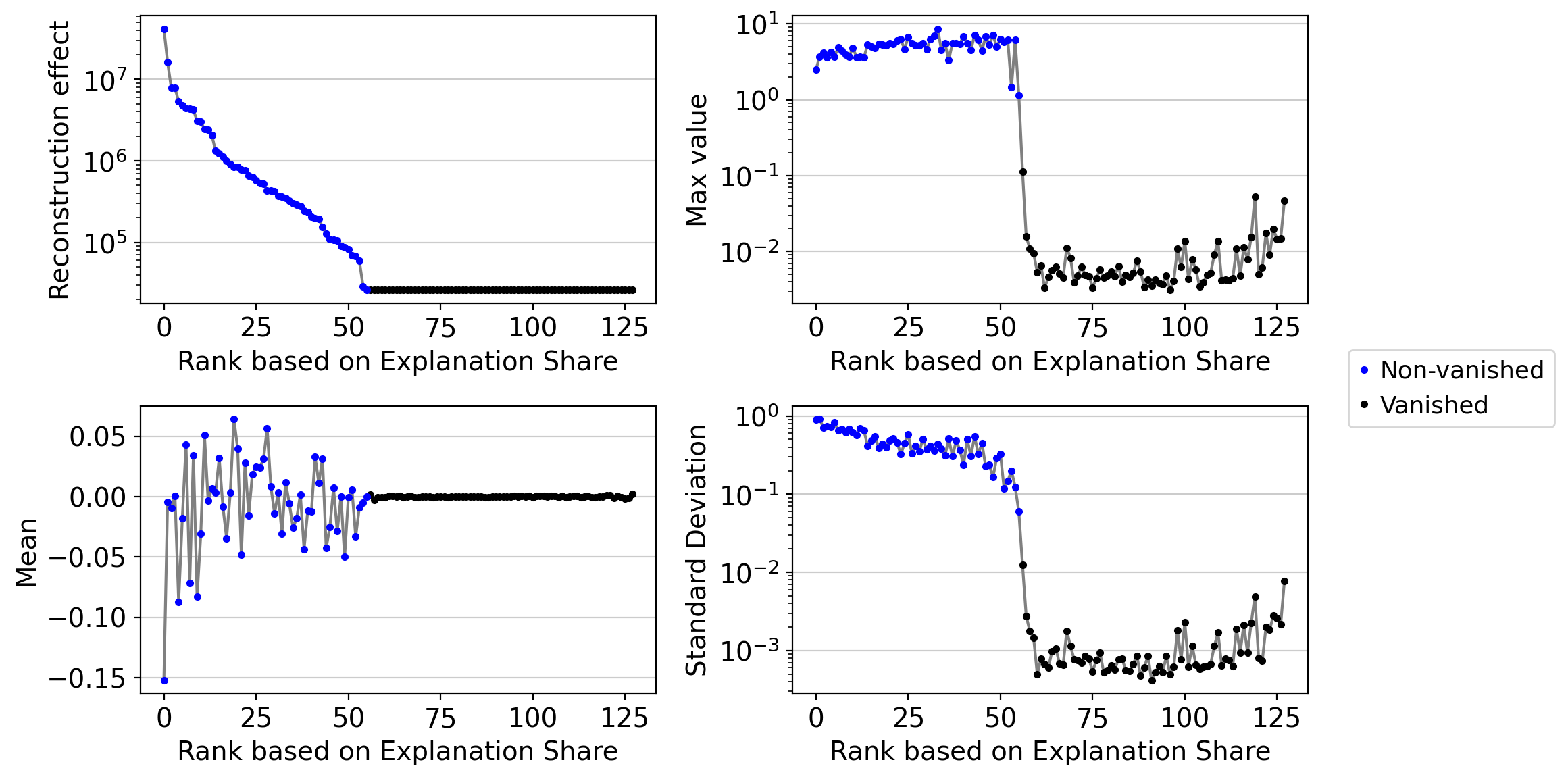
embed.var.sort_values("reconstruction_effect", ascending=False)[:5]
| original_dim_id | reconstruction_effect | order | max_value | mean | min | max | std | std_abs | title | vanished | vanished_positive_direction | vanished_negative_direction | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 120 | 120 | 41632488.0 | 0 | 2.483549 | -0.152309 | -2.483549 | 2.024353 | 0.904841 | 0.471779 | DR 1 | False | False | False |
| 49 | 49 | 16319570.0 | 1 | 3.713669 | -0.004452 | -3.713669 | 1.571943 | 0.909893 | 0.590519 | DR 2 | False | False | False |
| 17 | 17 | 7838704.5 | 2 | 4.173703 | -0.009392 | -4.173703 | 1.330191 | 0.713974 | 0.602237 | DR 3 | False | False | False |
| 55 | 55 | 7814997.0 | 3 | 3.620830 | 0.000510 | -3.620830 | 1.461988 | 0.733992 | 0.603089 | DR 4 | False | False | False |
| 12 | 12 | 5391969.5 | 4 | 4.273345 | -0.087206 | -4.273345 | 1.702258 | 0.728451 | 0.522156 | DR 5 | False | False | False |
drvi.utils.pl.plot_latent_dimension_stats(embed, ncols=2)
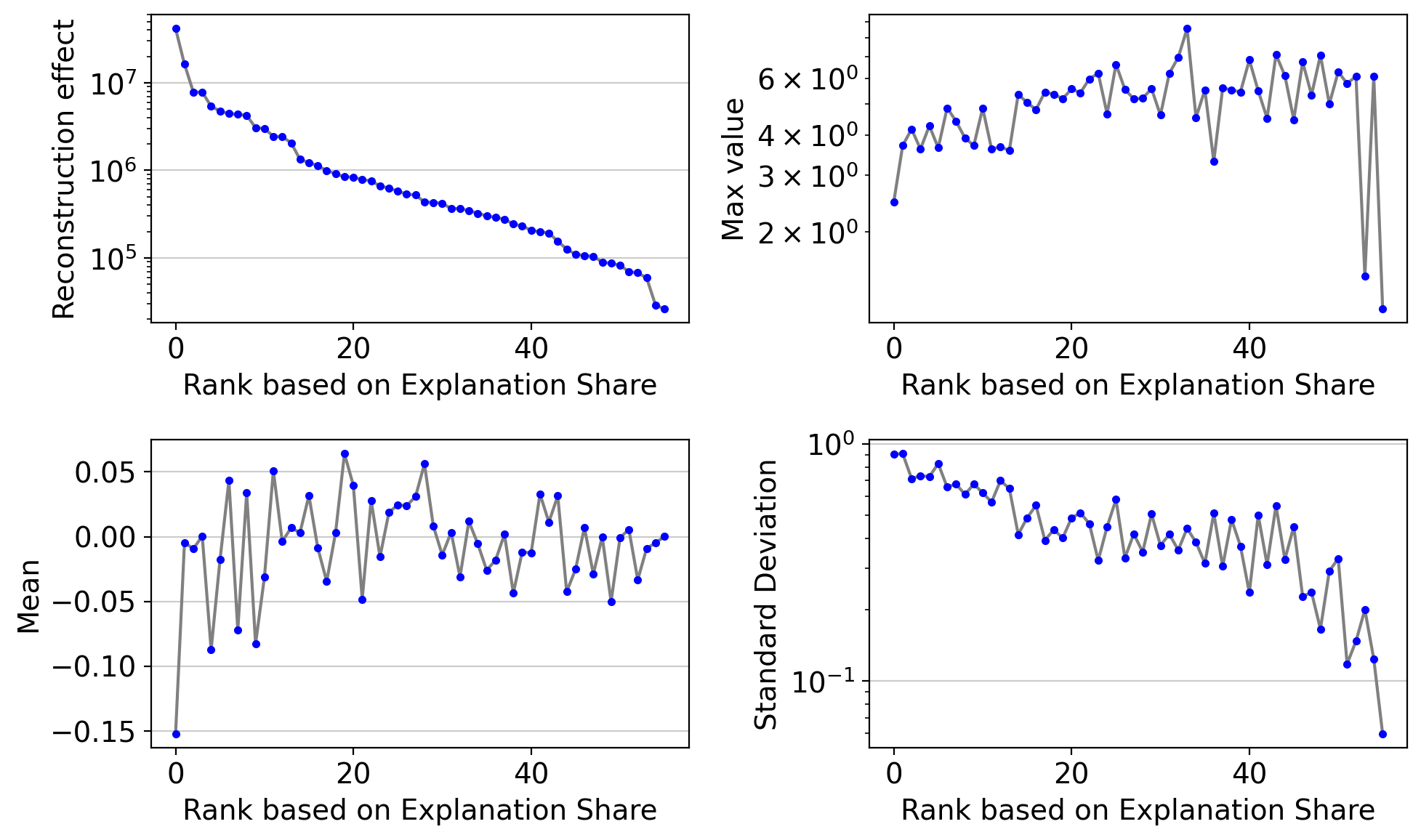
The same plot after removing vanished (inactive) dimensions:
drvi.utils.pl.plot_latent_dimension_stats(embed, ncols=2, remove_vanished=True)
Plot latent dimensions#
By default vanished dimensions are not plotted.
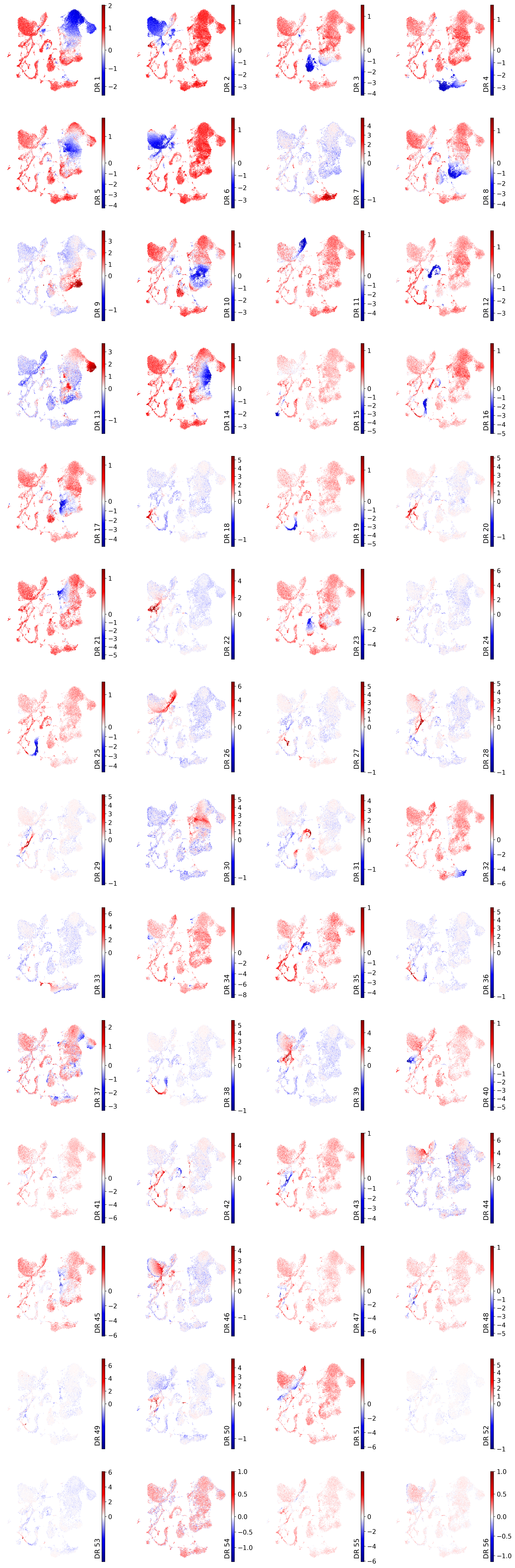
On the latent UMAP#
drvi.utils.pl.plot_latent_dims_in_umap(embed)
As a heatmap#
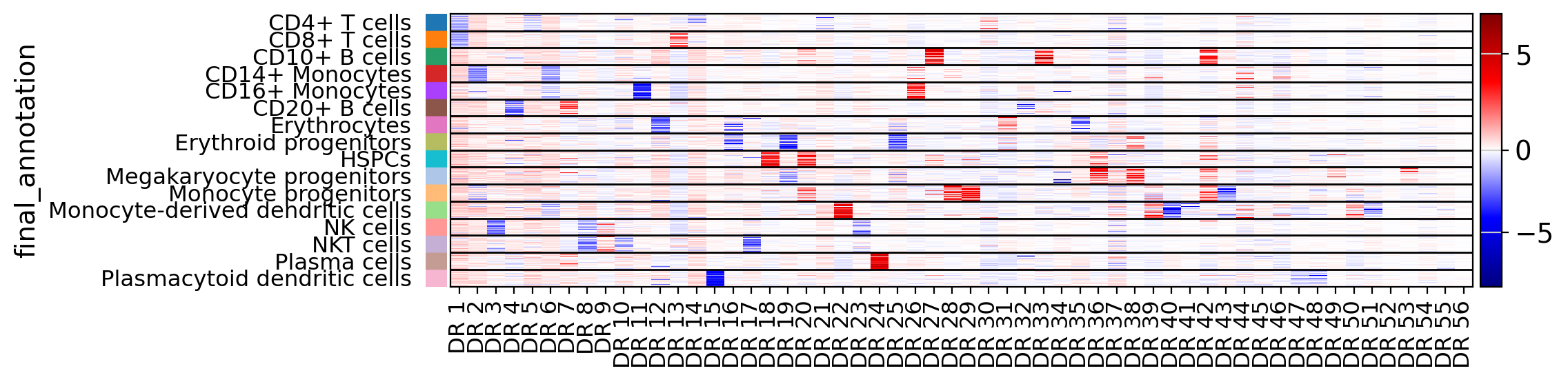
Heatmaps are useful to relate latent dimensions to known categories of the data. Dimensions can optionally be sorted by the category they are most relevant to.
drvi.utils.pl.plot_latent_dims_in_heatmap(embed, "final_annotation")
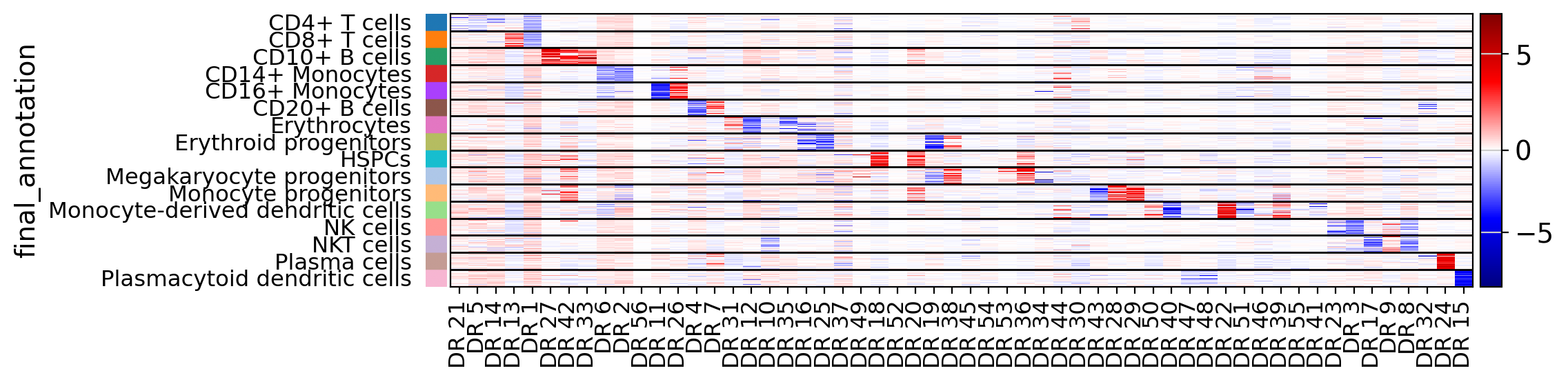
drvi.utils.pl.plot_latent_dims_in_heatmap(embed, "final_annotation", sort_by_categorical=True)
Interpretability#
The per-gene scores were already computed above and live in embed.varm.
embed.varm
AxisArrays with keys: OOD_min_possible_positive, OOD_min_possible_negative, OOD_max_possible_positive, OOD_max_possible_negative, OOD_combined_positive, OOD_combined_negative, IND_max_positive, IND_max_negative, IND_linear_weighted_mean_positive, IND_linear_weighted_mean_negative, IND_exp_weighted_mean_positive, IND_exp_weighted_mean_negative
Out-of-distribution (OOD) scores#
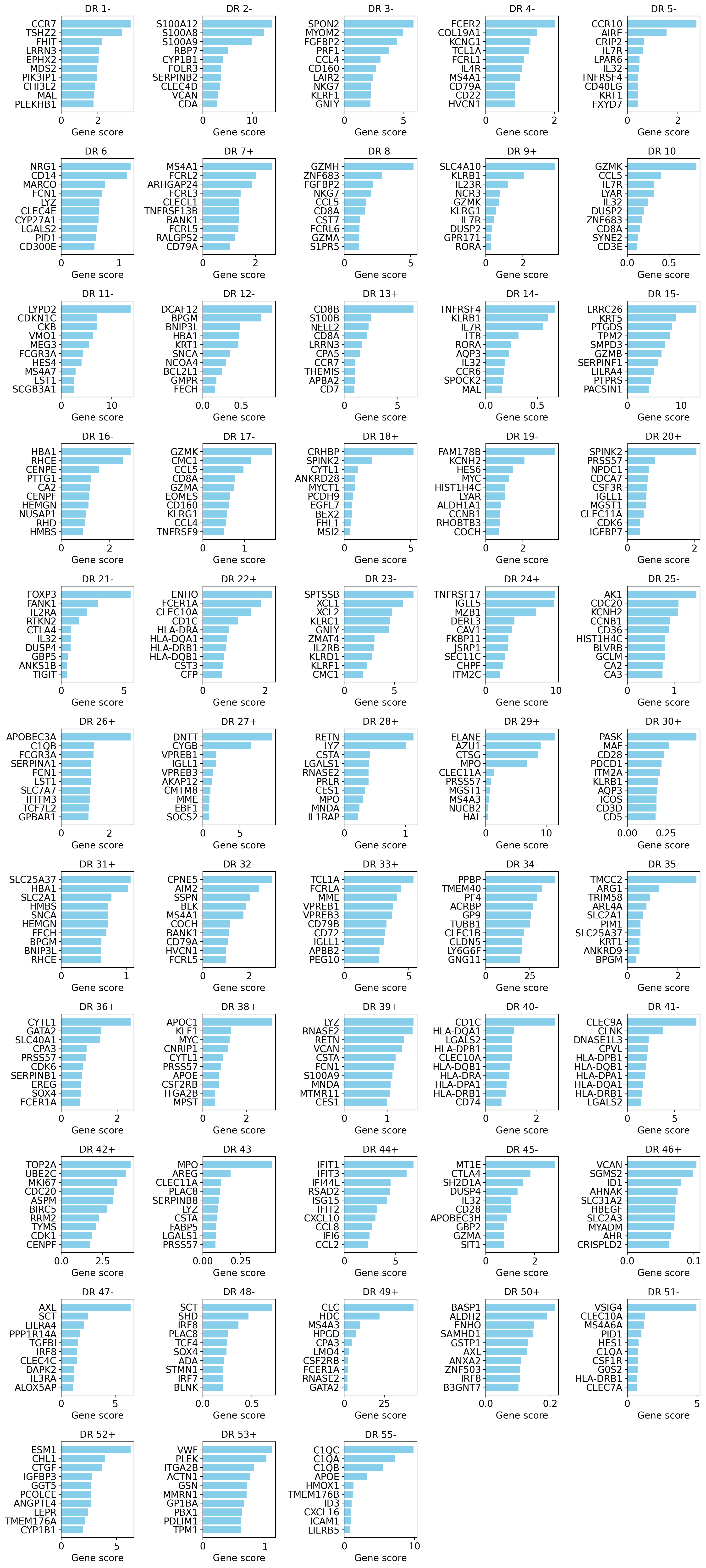
The default OOD_combined score combines the maximum effect and the specificity of each gene per dimension, which makes it our suggested score for finding the most specific genes of a program. If human-readable gene symbols live in a column of adata.var, pass it as gene_symbols=....
All scores can be obtained as a DataFrame (genes × dimensions). This includes all non-vanished dimensions / directions.
gene_scores_df = model.get_interpretability_scores(embed, adata)
gene_scores_df.iloc[:10, :10]
| title | DR 1+ | DR 1- | DR 2+ | DR 2- | DR 3+ | DR 3- | DR 4+ | DR 4- | DR 5+ | DR 5- |
|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||
| TCL1A | 2.412687e-10 | 0.001456 | 5.522654e-11 | 0.000408 | 0.000000e+00 | 0.000791 | 5.305386e-12 | 1.257571 | 1.185880e-10 | 0.000328 |
| IGLL5 | 8.337035e-13 | 0.000098 | 0.000000e+00 | 0.000146 | 0.000000e+00 | 0.000009 | 0.000000e+00 | 0.109184 | 0.000000e+00 | 0.000045 |
| PTGDS | 0.000000e+00 | 0.000579 | 3.385341e-12 | 0.000223 | 6.038034e-12 | 0.572757 | 6.456958e-09 | 0.000003 | 0.000000e+00 | 0.000644 |
| GZMB | 7.221287e-09 | 0.003130 | 4.590574e-09 | 0.000746 | 2.380274e-09 | 1.358150 | 4.744122e-08 | 0.000223 | 9.215202e-10 | 0.000249 |
| PPBP | 1.630017e-10 | 0.000183 | 1.078964e-09 | 0.000427 | 1.198929e-08 | 0.000013 | 6.093008e-09 | 0.000033 | 1.941316e-09 | 0.000007 |
| CD79A | 4.466629e-11 | 0.003331 | 3.197026e-08 | 0.000626 | 2.514043e-08 | 0.000338 | 1.575447e-10 | 0.856043 | 3.677643e-10 | 0.000492 |
| FGFBP2 | 2.082743e-10 | 0.004022 | 7.083195e-10 | 0.000607 | 1.540659e-09 | 4.489642 | 7.033426e-10 | 0.001020 | 3.321929e-10 | 0.000340 |
| FCGR3A | 3.294372e-08 | 0.003072 | 1.656720e-09 | 0.002539 | 9.794753e-11 | 0.732972 | 5.033220e-09 | 0.000210 | 1.163810e-08 | 0.000223 |
| GNLY | 6.625820e-09 | 0.012571 | 1.878915e-09 | 0.002714 | 2.870239e-09 | 2.217784 | 2.881133e-08 | 0.002135 | 1.297420e-09 | 0.002112 |
| GZMH | 1.557257e-10 | 0.006391 | 8.314298e-10 | 0.000970 | 1.515572e-09 | 1.020924 | 8.565166e-10 | 0.000654 | 1.296662e-09 | 0.000201 |
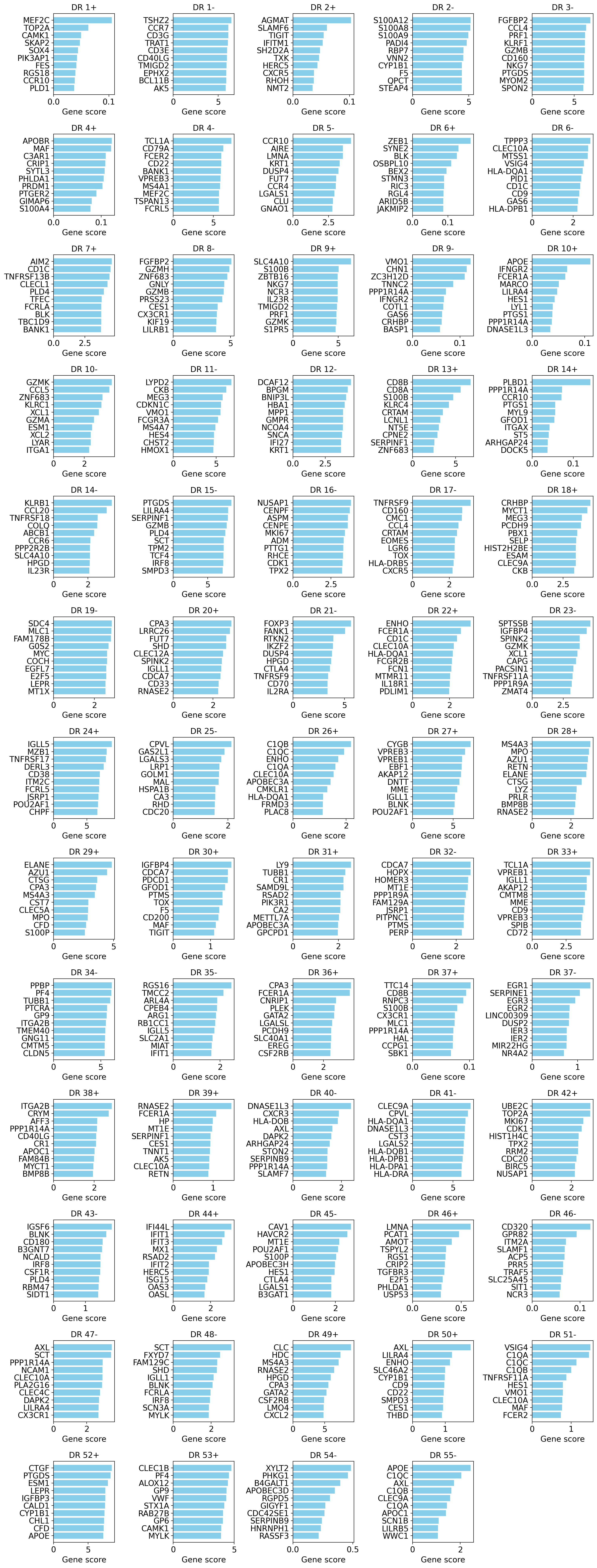
drvi.utils.pl.plot_interpretability_scores(gene_scores_df, score_threshold=0.1)
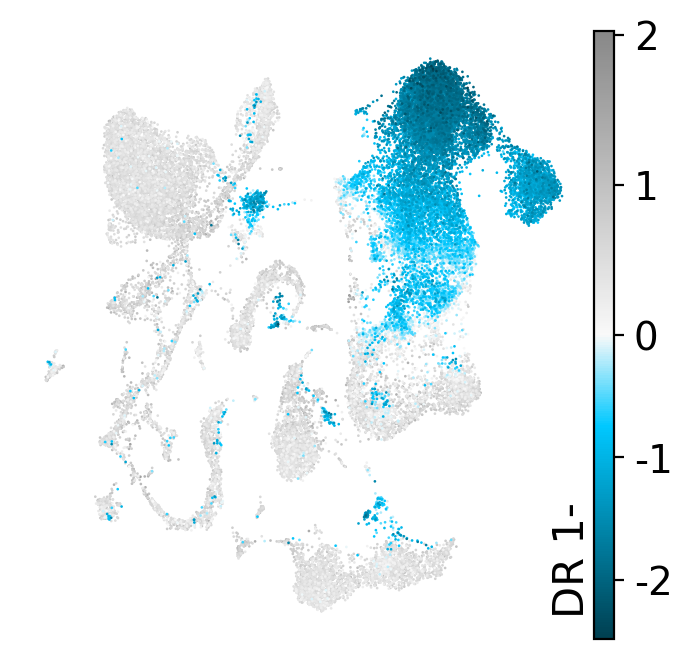
We can dig into individual dimensions: visualize a dimension on the UMAP (directional, so + and - are shown separately) and color the cells by its top genes. The exact dimension titles vary with the run and initialization.
# copy the latent UMAP onto the original (gene-level) AnnData
adata.obsm["X_drvi_umap"] = embed[adata.obs.index].obsm["X_umap"]
# We only show a few interpretable dimensions
for dim_title in gene_scores_df.columns[:4]:
if gene_scores_df[dim_title].max() < 0.1:
continue
top_genes = gene_scores_df[dim_title].sort_values(ascending=False).index.to_list()[:4]
print(dim_title)
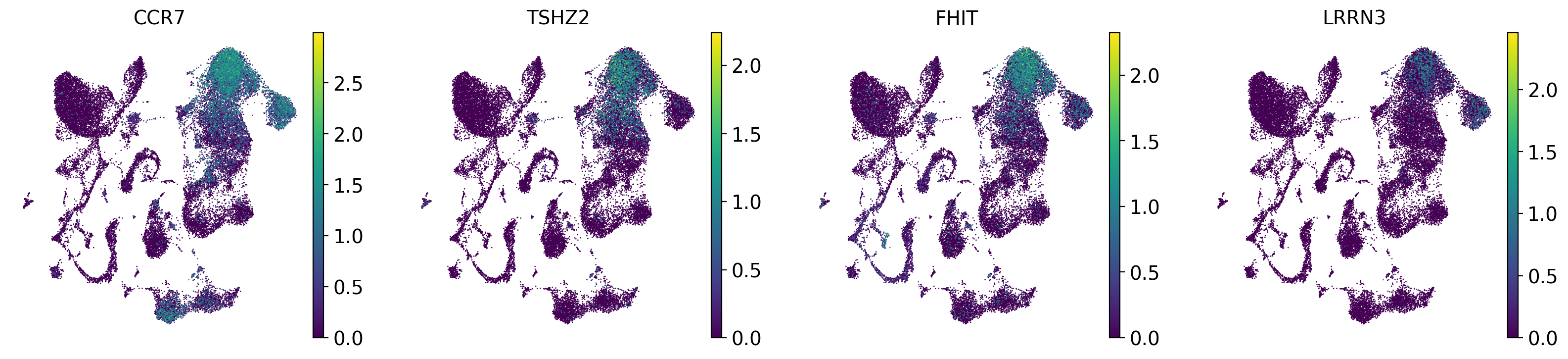
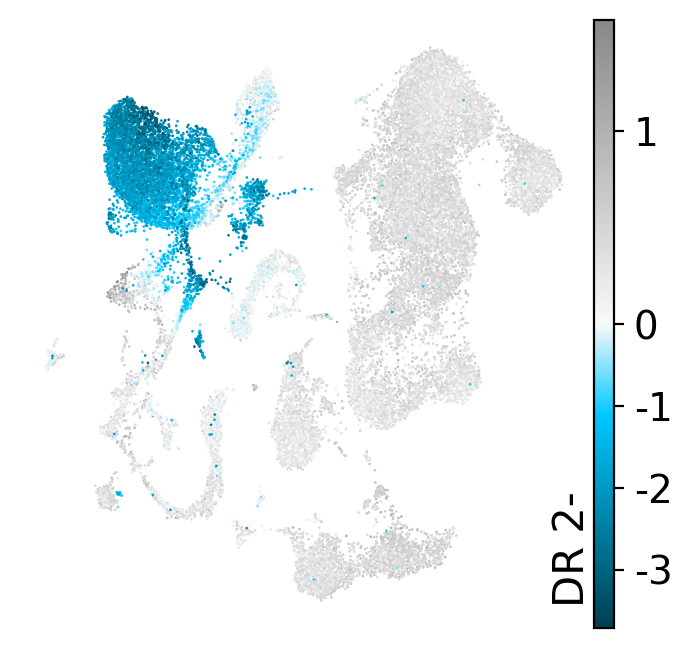
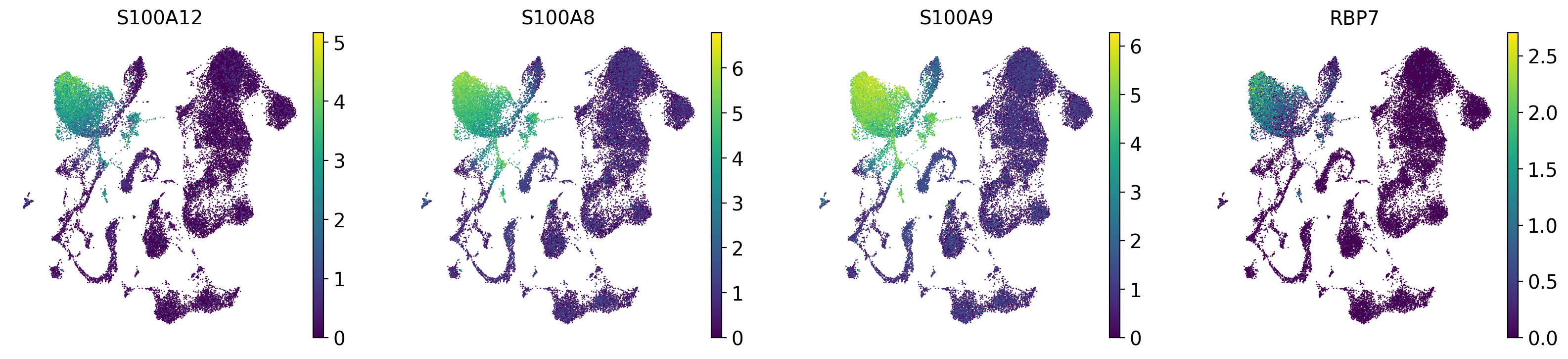
drvi.utils.pl.plot_latent_dims_in_umap(embed, dim_subset=[dim_title], directional=True)
sc.pl.embedding(adata, "X_drvi_umap", color=top_genes, frameon=False)
DR 1-
DR 2-
In-distribution (IND) scores#
The IND scores average each dimension’s effect on each gene over all cells. Because genes are not filtered for uniqueness, broadly affected genes also keep high scores, giving a complete view of how each factor influences the transcriptome.
gene_scores_df_ind = model.get_interpretability_scores(
embed, adata, key="IND_linear_weighted_mean"
)
gene_scores_df_ind.iloc[:10, :10]
| title | DR 1+ | DR 1- | DR 2+ | DR 2- | DR 3+ | DR 3- | DR 4+ | DR 4- | DR 5+ | DR 5- |
|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||
| TCL1A | 0.005971 | 2.497063 | 0.001167 | 1.160384 | 0.001384 | 2.114294 | 0.000416 | 7.239596 | 0.002750 | 1.008309 |
| IGLL5 | 0.000448 | 2.196086 | 0.000176 | 0.860792 | 0.000197 | 0.970224 | 0.000064 | 4.572513 | 0.000302 | 1.080758 |
| PTGDS | 0.000874 | 2.526320 | 0.000307 | 1.285834 | 0.000281 | 6.163900 | 0.005660 | 0.796921 | 0.000019 | 1.724263 |
| GZMB | 0.006450 | 2.521758 | 0.004755 | 1.187824 | 0.000510 | 6.265604 | 0.013632 | 1.810183 | 0.001139 | 0.799658 |
| PPBP | 0.004878 | 1.948129 | 0.000111 | 1.741534 | 0.012278 | 0.778982 | 0.010038 | 1.145155 | 0.004759 | 0.348709 |
| CD79A | 0.000977 | 3.127899 | 0.006475 | 1.488011 | 0.003377 | 1.750263 | 0.000041 | 6.236791 | 0.001201 | 0.870174 |
| FGFBP2 | 0.000994 | 3.066175 | 0.000957 | 1.183964 | 0.000028 | 6.899695 | 0.000781 | 1.997372 | 0.000521 | 0.898678 |
| FCGR3A | 0.009571 | 2.601117 | 0.002131 | 1.532953 | 0.000113 | 6.004457 | 0.007521 | 1.132685 | 0.005178 | 0.658718 |
| GNLY | 0.002227 | 2.617745 | 0.000389 | 1.504693 | 0.000078 | 5.977234 | 0.005475 | 1.900974 | 0.000716 | 0.890538 |
| GZMH | 0.001197 | 2.238113 | 0.000904 | 1.064236 | 0.000088 | 5.464810 | 0.002496 | 1.482707 | 0.001472 | 0.594975 |
drvi.utils.pl.plot_interpretability_scores(gene_scores_df_ind)
Identification of programs#
Once the top relevant genes of a dimension are known, the corresponding biological program can be identified using external information such as existing annotations, expert examination, gene-set enrichment analysis (GSEA), the literature, or automated annotation tools. Because this supervised information is never given to the model, the quality of the discovered signatures is neither affected nor biased by it — unidentified dimensions with high gene scores are promising candidates for further investigation.
More resources#
This notebook covered the core DRVI workflow. For more:
More tutorials — the DRVI documentation hosts additional tutorials (e.g. identification of rare cell types and mapping query data onto a DRVI reference): https://drvi.readthedocs.io/latest/tutorials/
drvi-pypackage — beyond the plotting helpers used here, the companiondrvi-pypackage (pip install drvi-py) provides more utility functions for latent-space analysis. See the API documentation for the full set of tools and plotting functions.