8. Introduction to gimVI¶
8.1. Impute missing genes in Spatial Data from Sequencing Data¶
[1]:
import sys
sys.path.append("../../")
sys.path.append("../")
[2]:
def allow_notebook_for_test():
print("Testing the gimvi notebook")
test_mode = False
save_path = "data/"
def if_not_test_else(x, y):
if not test_mode:
return x
else:
return y
if not test_mode:
save_path = "../../data"
[3]:
from scvi.dataset import (
PreFrontalCortexStarmapDataset,
FrontalCortexDropseqDataset,
SmfishDataset,
CortexDataset,
)
from scvi.models import JVAE, Classifier
from scvi.inference import JVAETrainer
import notebooks.utils.gimvi_tutorial as gimvi_utils
INFO:hyperopt.utils:Failed to load dill, try installing dill via "pip install dill" for enhanced pickling support.
INFO:hyperopt.fmin:Failed to load dill, try installing dill via "pip install dill" for enhanced pickling support.
INFO:hyperopt.mongoexp:Failed to load dill, try installing dill via "pip install dill" for enhanced pickling support.
[4]:
import numpy as np
import copy
8.2. Load two datasets: one with spatial data, one from sequencing¶
Here we load: - Cortex: a scRNA-seq dataset of 3,005 mouse somatosensory cortex cells (Zeisel et al., 2015) - osmFISH: a smFISH dataset of 4,462 cells and 33 genes from the same tissue (Codeluppi et al., 2018)
[5]:
data_spatial = SmfishDataset(save_path=save_path)
data_seq = CortexDataset(
save_path=save_path, total_genes=None
)
# make sure gene names have the same case
data_spatial.make_gene_names_lower()
data_seq.make_gene_names_lower()
# filters genes by gene_names
data_seq.filter_genes_by_attribute(data_spatial.gene_names)
if test_mode:
data_seq = data_spatial
INFO:scvi.dataset.dataset:File ../../data/osmFISH_SScortex_mouse_all_cell.loom already downloaded
INFO:scvi.dataset.smfish:Loading smFISH dataset
../../scvi/dataset/dataset.py:1276: RuntimeWarning: divide by zero encountered in log
log_counts = np.log(data.sum(axis=1))
/home/achille/miniconda3/envs/scvi-update/lib/python3.7/site-packages/numpy/core/_methods.py:117: RuntimeWarning: invalid value encountered in subtract
x = asanyarray(arr - arrmean)
INFO:scvi.dataset.dataset:Computing the library size for the new data
INFO:scvi.dataset.dataset:Downsampled from 6471 to 4530 cells
INFO:scvi.dataset.dataset:Remapping labels to [0,N]
INFO:scvi.dataset.dataset:Remapping batch_indices to [0,N]
INFO:scvi.dataset.dataset:File ../../data/expression.bin already downloaded
INFO:scvi.dataset.cortex:Loading Cortex data
INFO:scvi.dataset.cortex:Finished preprocessing Cortex data
INFO:scvi.dataset.dataset:Remapping labels to [0,N]
INFO:scvi.dataset.dataset:Remapping batch_indices to [0,N]
FrontalCortexDropseq: a scRNA-seq dataset of 71,639 mouse frontal cortex cells (Saunders et al., 2018)
PreFrontalCortexStarmap: a starMAP dataset of 3,704 cells and 166 genes from the mouse pre-frontal cortex (Wang et al., 2018)
[6]:
# data_spatial = PreFrontalCortexStarmapDataset(save_path=save_path)
# data_seq = FrontalCortexDropseqDataset(
# save_path=save_path, genes_to_keep=data_spatial.gene_names
# )
# data_seq.subsample_cells(5000)
Hide some genes in the osFISH dataset to score the imputation
[7]:
data_seq.filter_cells_by_count(1)
data_spatial.filter_cells_by_count(1)
INFO:scvi.dataset.dataset:Computing the library size for the new data
INFO:scvi.dataset.dataset:Downsampled from 3005 to 2996 cells
INFO:scvi.dataset.dataset:Computing the library size for the new data
INFO:scvi.dataset.dataset:Downsampled from 4530 to 4530 cells
[8]:
train_size = 0.8
gene_names_rnaseq = data_seq.gene_names
np.random.seed(0)
n_genes = len(gene_names_rnaseq)
gene_ids_train = sorted(
np.random.choice(range(n_genes), int(n_genes * train_size), False)
)
gene_ids_test = sorted(set(range(n_genes)) - set(gene_ids_train))
gene_names_fish = gene_names_rnaseq[gene_ids_train]
# Create copy of the fish dataset with hidden genes
data_spatial_partial = copy.deepcopy(data_spatial)
data_spatial_partial.filter_genes_by_attribute(gene_names_fish)
data_spatial_partial.batch_indices += data_seq.n_batches
INFO:scvi.dataset.dataset:Downsampling from 33 to 26 genes
INFO:scvi.dataset.dataset:Computing the library size for the new data
INFO:scvi.dataset.dataset:Filtering non-expressing cells.
INFO:scvi.dataset.dataset:Computing the library size for the new data
INFO:scvi.dataset.dataset:Downsampled from 4530 to 4530 cells
Configure the Joint Model The joint model can take multiple datasets with potentially different observed genes. All dataset will be encoded and decoded with the union of all genes. It requires: - The gene mappings from each dataset to the common decoded vector: * Eg: dataset1 has genes [‘a’, ‘b’] and dataset2 has genes [‘b’, ‘c’], then a possible output can be [‘b’, ‘a’, ‘c’] such that the mappings are [1, 0] and [0, 2] * Usually, if the genes of dataset2 are included in dataset1, it is way more efficient to keep the order of dataset1 in the output and use ``slice(None)`` as a mapping for dataset1
The number of inputs (ie) number of genes in each dataset
The distributions to use for the generative process: usually scRNA-seq is modelled with ZINB (because of technical dropout) and FISH with NB or even Poisson
Whether to model the library size with a latent variable or use the observed value
[9]:
datasets = [data_seq, data_spatial_partial]
generative_distributions = ["zinb", "nb"]
gene_mappings = [slice(None), np.array(gene_ids_train)]
n_inputs = [d.nb_genes for d in datasets]
total_genes = data_seq.nb_genes
n_batches = sum([d.n_batches for d in datasets])
model_library_size = [True, False]
n_latent = 8
kappa = 1
[10]:
import torch
torch.manual_seed(0)
model = JVAE(
n_inputs,
total_genes,
gene_mappings,
generative_distributions,
model_library_size,
n_layers_decoder_individual=0,
n_layers_decoder_shared=0,
n_layers_encoder_individual=1,
n_layers_encoder_shared=1,
dim_hidden_encoder=64,
dim_hidden_decoder_shared=64,
dropout_rate_encoder=0.2,
dropout_rate_decoder=0.2,
n_batch=n_batches,
n_latent=n_latent,
)
discriminator = Classifier(n_latent, 32, 2, 3, logits=True)
trainer = JVAETrainer(model, discriminator, datasets, 0.95, frequency=1, kappa=kappa)
[11]:
n_epochs = if_not_test_else(200, 1)
trainer.train(n_epochs=n_epochs)
training: 100%|██████████| 200/200 [04:24<00:00, 1.27s/it]
[12]:
gimvi_utils.plot_umap(trainer)

[13]:
gimvi_utils.imputation_score(trainer, data_spatial, gene_ids_test, True)
[13]:
0.25089044336342275
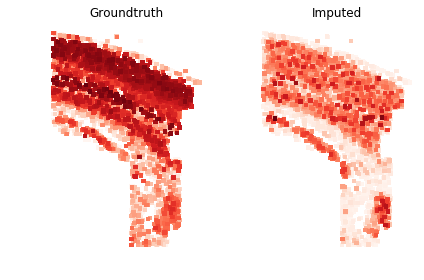
8.2.2. Inspect classification accuracy (we expect a uniform matrix)¶
If the matrix is diagonal, the kappa needs to be scaled up to ensure mixing.
[15]:
discriminator_classification = trainer.get_discriminator_confusion()
discriminator_classification
[15]:
array([[0.5616336 , 0.43836704],
[0.41276518, 0.58723474]], dtype=float32)
[16]:
import pandas as pd
results = pd.DataFrame(
trainer.get_loss_magnitude(),
index=["reconstruction", "kl_divergence", "discriminator"],
columns=["Sequencing", "Spatial"],
)
results.columns.name = "Dataset"
results.index.name = "Loss"
results
[16]:
| Dataset | Sequencing | Spatial |
|---|---|---|
| Loss | ||
| reconstruction | 868.178913 | 1765.545235 |
| kl_divergence | 201.512057 | 206.783453 |
| discriminator | 24.937387 | 20.623563 |