CITE-seq analysis in R#
In this brief tutorial, we go over how to use scvi-tools functionality in R for analyzing CITE-seq data. We will closely follow the Bioconductor PBMC tutorial, using totalVI when appropriate.
This tutorial requires Reticulate. Please check out our installation guide for instructions on installing Reticulate and scvi-tools.
Loading and processing data with Bioconductor#
library(BiocFileCache)
bfc <- BiocFileCache(ask=FALSE)
exprs.data <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples/cell-vdj/3.1.0",
"vdj_v1_hs_pbmc3",
"vdj_v1_hs_pbmc3_filtered_feature_bc_matrix.tar.gz"))
untar(exprs.data, exdir=tempdir())
library(DropletUtils)
sce.pbmc <- read10xCounts(file.path(tempdir(), "filtered_feature_bc_matrix"))
sce.pbmc <- splitAltExps(sce.pbmc, rowData(sce.pbmc)$Type)
Pre-processing and quality control#
unfiltered <- sce.pbmc
library(scater)
is.mito <- grep("^MT-", rowData(sce.pbmc)$Symbol)
stats <- perCellQCMetrics(sce.pbmc, subsets=list(Mito=is.mito))
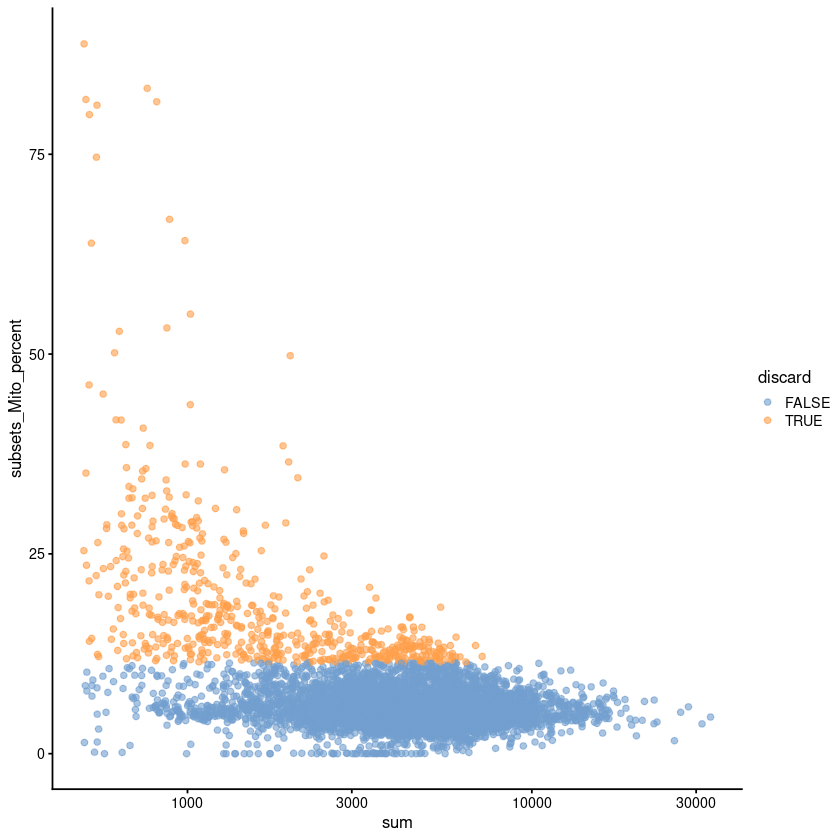
high.mito <- isOutlier(stats$subsets_Mito_percent, type="higher")
low.adt <- stats$`altexps_Antibody Capture_detected` < nrow(altExp(sce.pbmc))/2
discard <- high.mito | low.adt
sce.pbmc <- sce.pbmc[,!discard]
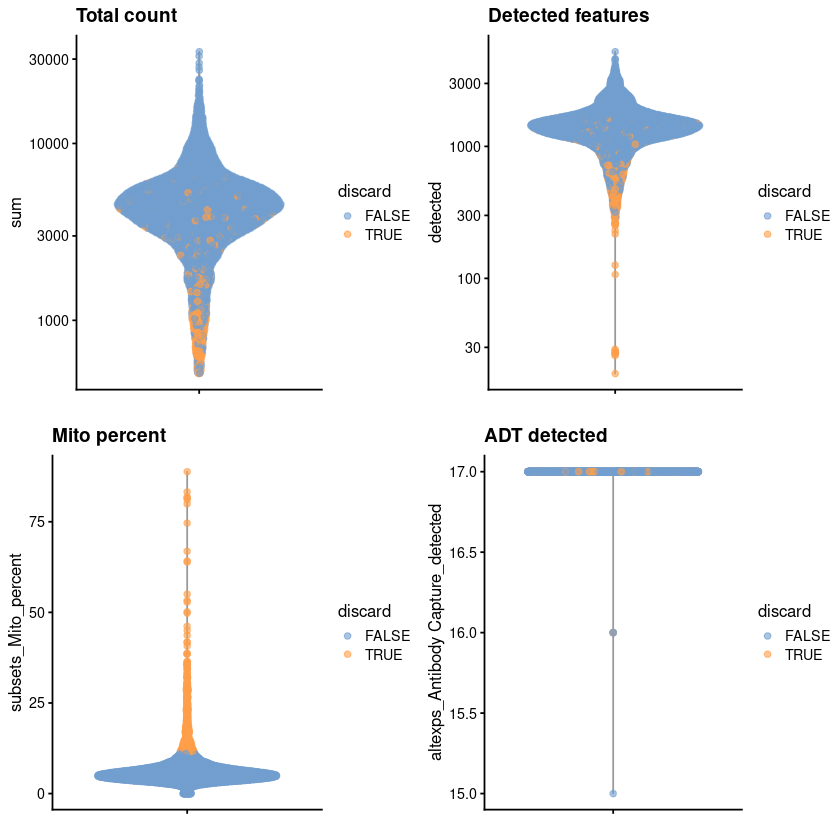
summary(high.mito)
Mode FALSE TRUE
logical 6660 571
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- discard
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
plotColData(unfiltered, y="altexps_Antibody Capture_detected",
colour_by="discard") + ggtitle("ADT detected"),
ncol=2
)
plotColData(unfiltered, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()
Normalization#
While we normalize the data here using standard Bioconductor practices, we will use the counts later for totalVI.
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.pbmc)
sce.pbmc <- computeSumFactors(sce.pbmc, cluster=clusters)
altExp(sce.pbmc) <- computeMedianFactors(altExp(sce.pbmc))
sce.pbmc <- logNormCounts(sce.pbmc, use_altexps=TRUE)
Data conversion (SCE -> AnnData)#
We use sceasy for conversion, and load the necessary Python packages for later.
library(reticulate)
library(sceasy)
library(anndata)
use_python("/home/adam/.pyenv/versions/3.9.7/bin/python", required = TRUE)
sc <- import("scanpy", convert = FALSE)
scvi <- import("scvi", convert = FALSE)
sys <- import ("sys", convert = FALSE)
We make two AnnData objects, one per modality, and then store the protein counts in the canonical location for scvi-tools.
adata <- convertFormat(sce.pbmc, from="sce", to="anndata", main_layer="counts", drop_single_values=FALSE)
adata_protein <- convertFormat(altExp(sce.pbmc), from="sce", to="anndata", main_layer="counts", drop_single_values=FALSE)
adata$obsm["protein"] <- adata_protein$to_df()
adata
AnnData object with n_obs × n_vars = 6660 × 33538
obs: 'Sample', 'Barcode', 'sizeFactor'
var: 'ID', 'Symbol', 'Type'
obsm: 'protein'
Run totalVI for dimensionality reduction#
totalVI will output a low-dimensional representation of cells that captures information from both the RNA and protein. Here we show how to use totalVI for only dimensionality reduction, though totalVI can perform other tasks that are shown in the Python-based tutorials. The intention here is to provide some examples of how to use totalVI from R.
scvi$model$TOTALVI$setup_anndata(adata, protein_expression_obsm_key="protein")
None
None
vae <- scvi$model$TOTALVI(adata)
vae$train()
None
reducedDims(sce.pbmc) <- list(TOTALVI=py_to_r(vae$get_latent_representation()))
sce.pbmc <- runUMAP(sce.pbmc, dimred="TOTALVI")
sce.pbmc <- runTSNE(sce.pbmc, dimred="TOTALVI")
Clustering#
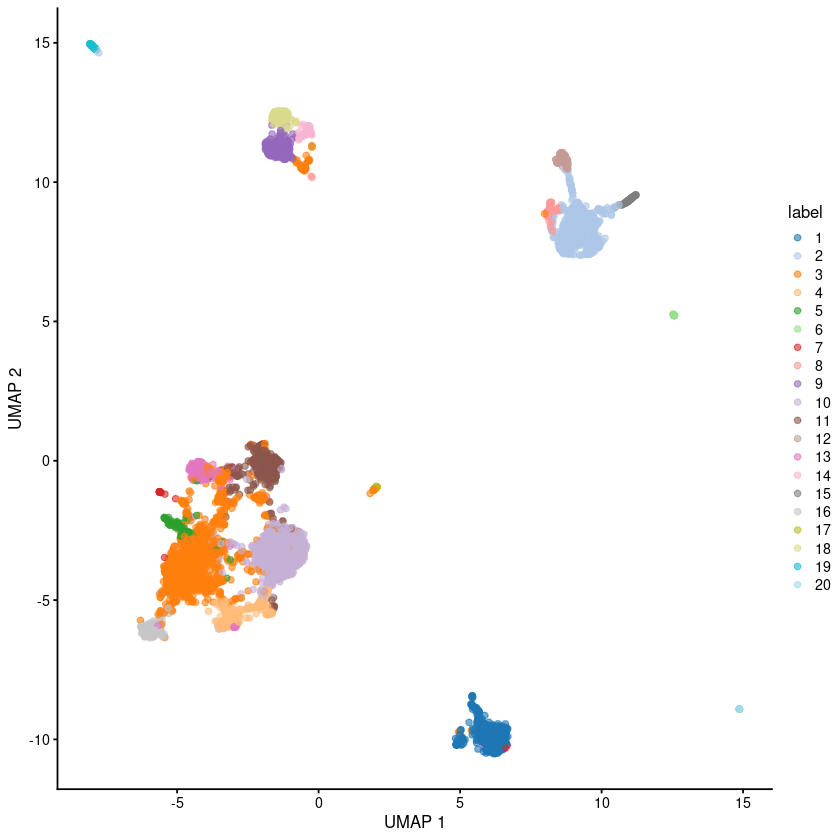
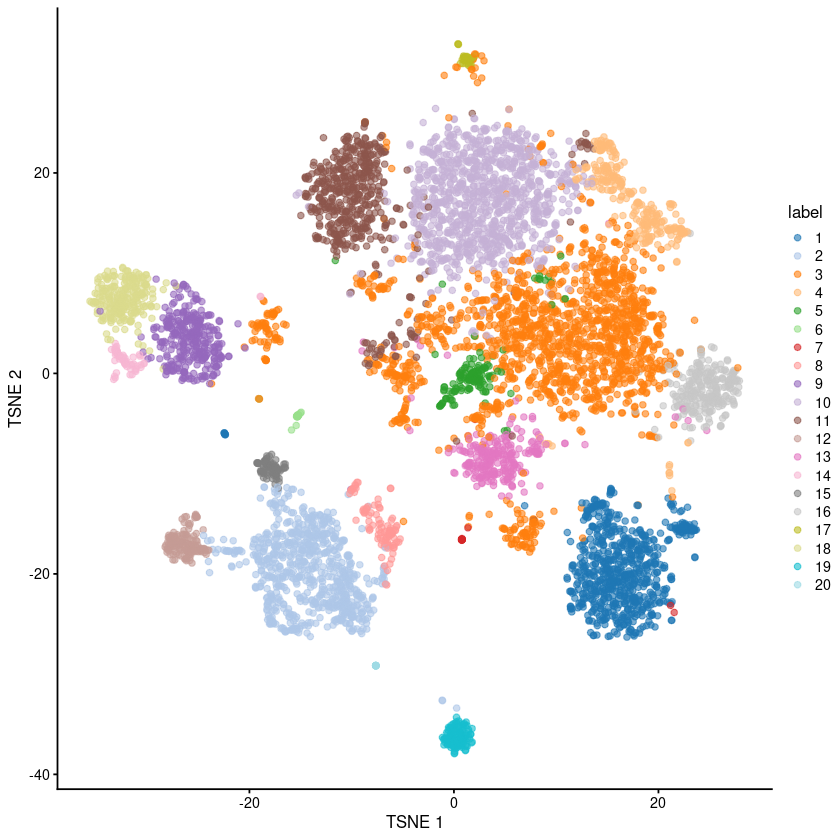
g <- buildSNNGraph(sce.pbmc, k=10, use.dimred = 'TOTALVI')
clust <- igraph::cluster_walktrap(g)$membership
colLabels(sce.pbmc) <- factor(clust)
plotUMAP(sce.pbmc, colour_by="label")
plotTSNE(sce.pbmc, colour_by="label")
sI <- sessionInfo()
sI$loadedOnly <- NULL
print(sI, locale=FALSE)
R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] anndata_0.7.5.3 sceasy_0.0.6
[3] reticulate_1.20 scran_1.21.3
[5] scater_1.21.3 ggplot2_3.3.5
[7] scuttle_1.3.1 DropletUtils_1.13.2
[9] SingleCellExperiment_1.15.2 SummarizedExperiment_1.23.4
[11] Biobase_2.53.0 GenomicRanges_1.45.0
[13] GenomeInfoDb_1.29.8 IRanges_2.27.2
[15] S4Vectors_0.31.3 BiocGenerics_0.39.2
[17] MatrixGenerics_1.5.4 matrixStats_0.60.1
[19] BiocFileCache_2.1.1 dbplyr_2.1.1