Topic Modeling with Amortized LDA#
In this tutorial, we will explore how to run the amortized Latent Dirichlet Allocation (LDA) model implementation in scvi-tools. LDA is a topic modelling method first introduced in the natural language processing field. By treating each cell as a document and each gene expression count as a word, we can carry over the method to the single-cell biology field.
Below, we will train the model over a dataset, plot the topics over a UMAP of the reference set, and inspect the topics for characteristic gene sets.
As an example, we use the PBMC 10K dataset from 10x Genomics.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
WARNING: Running pip as the 'root' user can result in broken permissions and conflicting behaviour with the system package manager, possibly rendering your system unusable. It is recommended to use a virtual environment instead: https://pip.pypa.io/warnings/venv. Use the --root-user-action option if you know what you are doing and want to suppress this warning.
[notice] A new release of pip is available: 25.0.1 -> 26.1.2
[notice] To update, run: pip install --upgrade pip
import os
import tempfile
import pandas as pd
import scanpy as sc
import scvi
import seaborn as sns
import torch
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.5.0
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Load and process data#
Load the 10x genomics PBMC dataset. Generally, it is good practice for LDA to remove ubiquitous genes, to prevent the model from modeling these genes as a separate topic. Here, we first filter out all mitochrondrial genes, then select the top 1000 variable genes with seurat_v3 method from the remaining genes.
adata_path = os.path.join(save_dir.name, "pbmc_10k_protein_v3.h5ad")
adata = sc.read(
adata_path,
backup_url="https://github.com/YosefLab/scVI-data/raw/master/pbmc_10k_protein_v3.h5ad?raw=true",
)
adata.layers["counts"] = adata.X.copy() # preserve counts
sc.pp.normalize_total(adata, target_sum=10e4)
sc.pp.log1p(adata)
adata.raw = adata # freeze the state in `.raw`
adata = adata[:, ~adata.var_names.str.startswith("MT-")]
sc.pp.highly_variable_genes(
adata, flavor="seurat_v3", layer="counts", n_top_genes=1000, subset=True
)
Create and fit AmortizedLDA model#
Here, we initialize and fit an AmortizedLDA model on the dataset. We pick 10 topics to model in this case.
n_topics = 10
scvi.model.AmortizedLDA.setup_anndata(adata, layer="counts")
model = scvi.model.AmortizedLDA(adata, n_topics=n_topics)
Note
By default we train with KL annealing which means the effective loss will generally not decrease steadily in the beginning. Our Pyro implementations present this train loss term as the elbo_train in the progress bar which is misleading. We plan on correcting this in the future.
model.train()
Visualizing learned topics#
By calling model.get_latent_representation(), the model will compute a Monte Carlo estimate of the topic proportions for each cell. Since we use a logistic-Normal distribution to approximate the Dirichlet distribution, the model cannot compute the analytic mean. The number of samples used to compute the latent representation can be configured with the optional argument n_samples.
topic_prop = model.get_latent_representation()
topic_prop.head()
| topic_0 | topic_1 | topic_2 | topic_3 | topic_4 | topic_5 | topic_6 | topic_7 | topic_8 | topic_9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||
| AAACCCAAGATTGTGA-1 | 0.000304 | 0.000242 | 0.000257 | 0.196104 | 0.786108 | 0.015958 | 0.000508 | 0.000278 | 0.000174 | 0.000066 |
| AAACCCACATCGGTTA-1 | 0.000089 | 0.000164 | 0.000163 | 0.000779 | 0.990523 | 0.006031 | 0.000245 | 0.001900 | 0.000051 | 0.000055 |
| AAACCCAGTACCGCGT-1 | 0.002808 | 0.000820 | 0.000659 | 0.436753 | 0.533710 | 0.004863 | 0.014890 | 0.002686 | 0.002536 | 0.000275 |
| AAACCCAGTATCGAAA-1 | 0.009934 | 0.004512 | 0.005277 | 0.005808 | 0.002674 | 0.005462 | 0.014656 | 0.362502 | 0.027906 | 0.561270 |
| AAACCCAGTCGTCATA-1 | 0.000981 | 0.000697 | 0.000440 | 0.001009 | 0.000624 | 0.000917 | 0.062733 | 0.419964 | 0.003604 | 0.509031 |
# Save topic proportions in obsm and obs columns.
adata.obsm["X_LDA"] = topic_prop
for i in range(n_topics):
adata.obs[f"LDA_topic_{i}"] = topic_prop[[f"topic_{i}"]]
Plot UMAP#
sc.tl.pca(adata, svd_solver="arpack")
sc.pp.neighbors(adata, n_pcs=30, n_neighbors=20)
sc.tl.umap(adata)
sc.tl.leiden(adata, key_added="leiden_scVI", resolution=0.8)
# Save UMAP to custom .obsm field.
adata.obsm["raw_counts_umap"] = adata.obsm["X_umap"].copy()
sc.pl.embedding(adata, "raw_counts_umap", color=["leiden_scVI"], frameon=False)
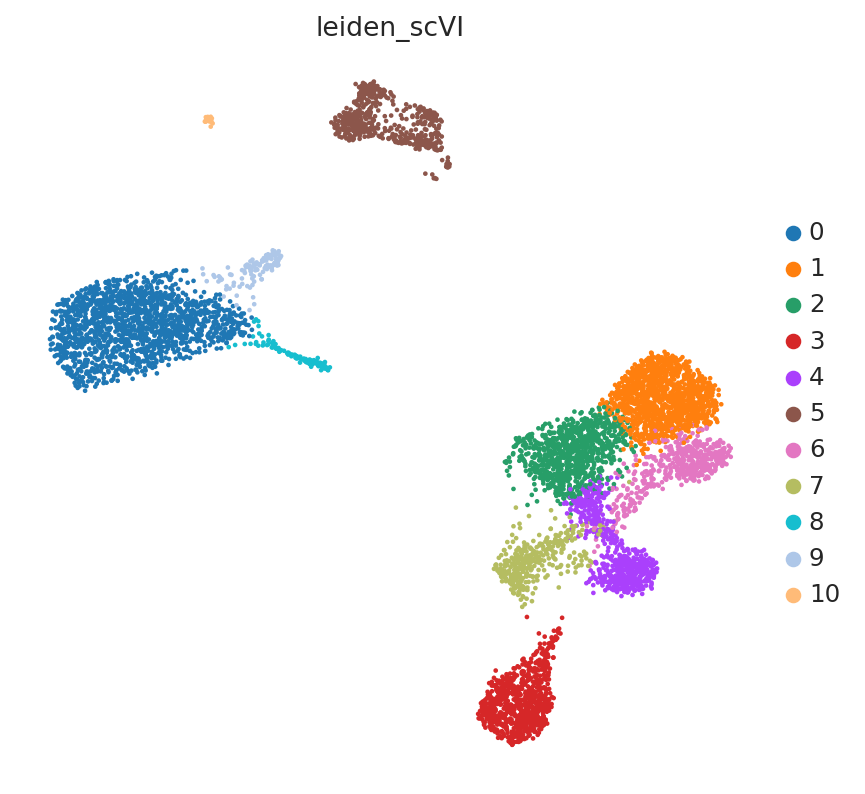
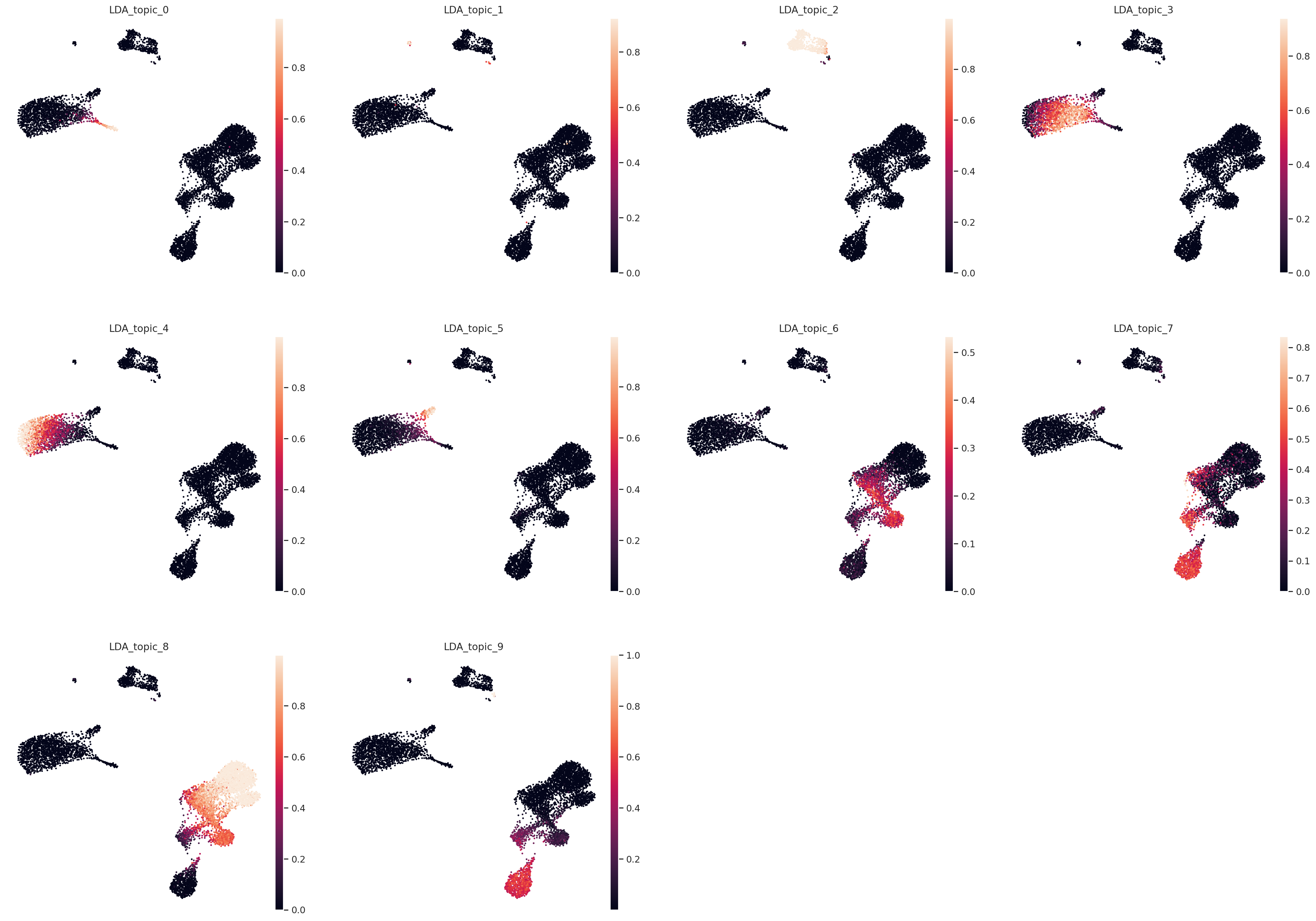
Color UMAP by topic proportions#
By coloring by UMAP by topic proportions, we find that the learned topics are generally dominant in cells close together in the UMAP space. In some cases, a topic is dominant in multiple clusters in the UMAP, which indicates similarilty between these clusters despite being far apart in the plot. This is not surprising considering that UMAP does not preserve local relationships beyond a certain threshold.
sc.pl.embedding(
adata,
"raw_counts_umap",
color=[f"LDA_topic_{i}" for i in range(n_topics)],
frameon=False,
)
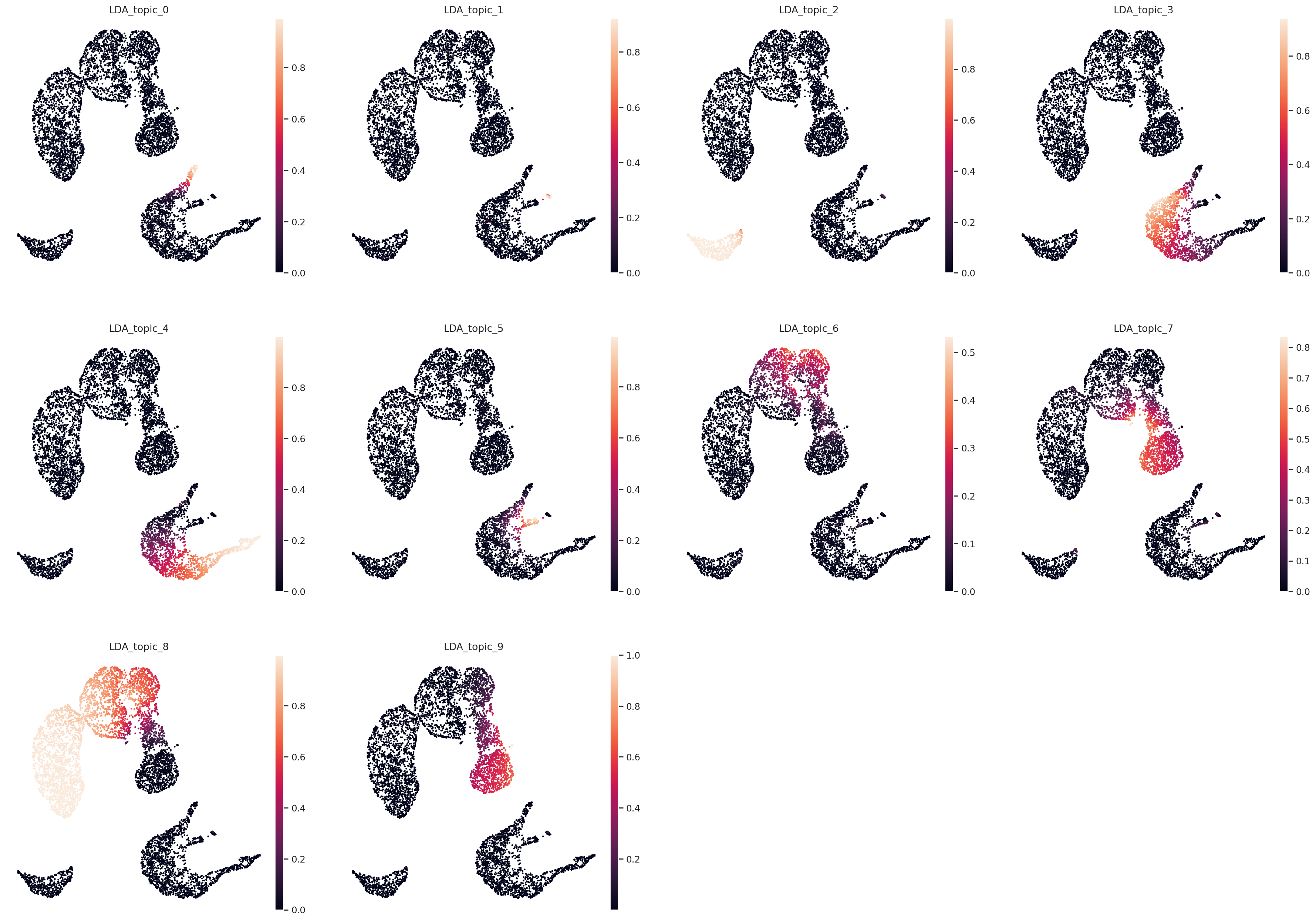
Plot UMAP in topic space#
sc.pp.neighbors(adata, use_rep="X_LDA", n_neighbors=20, metric="hellinger")
sc.tl.umap(adata)
# Save UMAP to custom .obsm field.
adata.obsm["topic_space_umap"] = adata.obsm["X_umap"].copy()
sc.pl.embedding(
adata,
"topic_space_umap",
color=[f"LDA_topic_{i}" for i in range(n_topics)],
frameon=False,
)
Find top genes per topic#
Similar to the topic proportions, model.get_feature_by_topic() returns a Monte Carlo estimate of the gene by topic matrix, which contains the proportion that a gene is weighted in each topic. This is also due to another approximation of the Dirichlet with a logistic-Normal distribution. We can inspect each topic in this matrix and sort by proportion allocated to each gene to determine top genes characterizing each topic.
feature_by_topic = model.get_feature_by_topic()
feature_by_topic.head()
| topic_0 | topic_1 | topic_2 | topic_3 | topic_4 | topic_5 | topic_6 | topic_7 | topic_8 | topic_9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| index | ||||||||||
| AL645608.8 | 0.000036 | 0.000023 | 0.000004 | 0.000001 | 2.783311e-06 | 0.000002 | 0.000006 | 0.000003 | 0.000001 | 0.000003 |
| HES4 | 0.000733 | 0.000028 | 0.000013 | 0.000007 | 8.456480e-06 | 0.000010 | 0.000011 | 0.000005 | 0.000007 | 0.000005 |
| ISG15 | 0.000720 | 0.000040 | 0.000413 | 0.000472 | 2.592755e-04 | 0.000133 | 0.001524 | 0.002357 | 0.001094 | 0.000327 |
| TNFRSF18 | 0.000004 | 0.000148 | 0.000035 | 0.000001 | 8.430371e-07 | 0.000002 | 0.000354 | 0.000023 | 0.000359 | 0.000138 |
| TNFRSF4 | 0.000004 | 0.000056 | 0.000007 | 0.000001 | 1.509900e-06 | 0.000004 | 0.000271 | 0.000035 | 0.000835 | 0.000026 |
rank_by_topic = pd.DataFrame()
for i in range(n_topics):
topic_name = f"topic_{i}"
topic = feature_by_topic[topic_name].sort_values(ascending=False)
rank_by_topic[topic_name] = topic.index
rank_by_topic[f"{topic_name}_prop"] = topic.values
rank_by_topic.head()
| topic_0 | topic_0_prop | topic_1 | topic_1_prop | topic_2 | topic_2_prop | topic_3 | topic_3_prop | topic_4 | topic_4_prop | topic_5 | topic_5_prop | topic_6 | topic_6_prop | topic_7 | topic_7_prop | topic_8 | topic_8_prop | topic_9 | topic_9_prop | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | FTL | 0.114418 | CD74 | 0.061230 | CD74 | 0.108184 | FTH1 | 0.059314 | S100A9 | 0.143670 | HLA-DRA | 0.085101 | S100A4 | 0.161333 | ACTB | 0.208307 | TMSB4X | 0.139633 | GNLY | 0.107086 |
| 1 | ACTB | 0.068806 | ACTB | 0.037643 | IGKC | 0.065732 | FTL | 0.057074 | S100A8 | 0.099610 | CD74 | 0.081020 | SH3BGRL3 | 0.042521 | TMSB4X | 0.177480 | TMSB10 | 0.095124 | NKG7 | 0.078788 |
| 2 | TMSB4X | 0.059545 | FTH1 | 0.028644 | HLA-DRA | 0.059559 | LYZ | 0.056565 | LYZ | 0.057241 | ACTB | 0.059655 | S100A6 | 0.039614 | TMSB10 | 0.079250 | ACTB | 0.094564 | IGKC | 0.077853 |
| 3 | FTH1 | 0.059212 | TMSB4X | 0.025981 | TMSB4X | 0.043953 | ACTB | 0.035131 | FTL | 0.042915 | HLA-DRB1 | 0.050423 | ANXA1 | 0.037930 | ACTG1 | 0.051750 | JUNB | 0.033868 | CCL5 | 0.054798 |
| 4 | S100A4 | 0.029395 | CYBA | 0.020130 | TMSB10 | 0.041685 | TMSB4X | 0.035055 | ACTB | 0.038939 | TMSB4X | 0.042672 | DUSP1 | 0.037028 | S100A4 | 0.047518 | FTL | 0.029890 | IGLC2 | 0.051152 |