Annotation with CellAssign#
Assigning single-cell RNA-seq data to known cell types#
CellAssign is a probabilistic model that uses prior knowledge of cell-type marker genes to annotate scRNA data into predefined cell types. Unlike other methods for assigning cell types, CellAssign does not require labeled single cell data and only needs to know whether or not each given gene is a marker of each cell type. The original paper and R code are linked below.
Code: https://github.com/Irrationone/cellassign
This notebook will demonstrate how to use CellAssign on follicular lymphoma and HGSC scRNA data.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
WARNING: Running pip as the 'root' user can result in broken permissions and conflicting behaviour with the system package manager, possibly rendering your system unusable. It is recommended to use a virtual environment instead: https://pip.pypa.io/warnings/venv. Use the --root-user-action option if you know what you are doing and want to suppress this warning.
[notice] A new release of pip is available: 25.0.1 -> 26.1.2
[notice] To update, run: pip install --upgrade pip
import os
import tempfile
import matplotlib.pyplot as plt
import pandas as pd
import scanpy as sc
import scvi
import seaborn as sns
import torch
from scvi.external import CellAssign
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.5.0
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
To demonstrate CellAssign, we use the data from the original publication, which we converted into h5ad format. The data are originally available from here:
https://zenodo.org/record/3372746
sce_follicular_path = os.path.join(save_dir.name, "sce_follicular.h5ad")
sce_hgsc_path = os.path.join(save_dir.name, "sce_hgsc.h5ad")
fl_celltype_path = os.path.join(save_dir.name, "fl_celltype.csv")
hgsc_celltype_path = os.path.join(save_dir.name, "hgsc_celltype.csv")
os.system("wget -q https://ndownloader.figshare.com/files/27458798 -O " + sce_follicular_path)
os.system("wget -q https://ndownloader.figshare.com/files/27458822 -O " + sce_hgsc_path)
os.system("wget -q https://ndownloader.figshare.com/files/27458828 -O " + hgsc_celltype_path)
os.system("wget -q https://ndownloader.figshare.com/files/27458831 -O " + fl_celltype_path)
0
Follicular Lymphoma Data#
Load follicular lymphoma data and marker gene matrix (see Supplementary Table 2 from the original paper).
follicular_adata = sc.read(sce_follicular_path)
fl_celltype_markers = pd.read_csv(fl_celltype_path, index_col=0)
follicular_adata.obs.index = follicular_adata.obs.index.astype("str")
follicular_adata.var.index = follicular_adata.var.index.astype("str")
follicular_adata.var_names_make_unique()
follicular_adata.obs_names_make_unique()
follicular_adata
AnnData object with n_obs × n_vars = 9156 × 33694
obs: 'Sample', 'dataset', 'patient', 'timepoint', 'progression_status', 'patient_progression', 'sample_barcode', 'is_cell_control', 'total_features_by_counts', 'log10_total_features_by_counts', 'total_counts', 'log10_total_counts', 'pct_counts_in_top_50_features', 'pct_counts_in_top_100_features', 'pct_counts_in_top_200_features', 'pct_counts_in_top_500_features', 'total_features_by_counts_endogenous', 'log10_total_features_by_counts_endogenous', 'total_counts_endogenous', 'log10_total_counts_endogenous', 'pct_counts_endogenous', 'pct_counts_in_top_50_features_endogenous', 'pct_counts_in_top_100_features_endogenous', 'pct_counts_in_top_200_features_endogenous', 'pct_counts_in_top_500_features_endogenous', 'total_features_by_counts_feature_control', 'log10_total_features_by_counts_feature_control', 'total_counts_feature_control', 'log10_total_counts_feature_control', 'pct_counts_feature_control', 'pct_counts_in_top_50_features_feature_control', 'pct_counts_in_top_100_features_feature_control', 'pct_counts_in_top_200_features_feature_control', 'pct_counts_in_top_500_features_feature_control', 'total_features_by_counts_mitochondrial', 'log10_total_features_by_counts_mitochondrial', 'total_counts_mitochondrial', 'log10_total_counts_mitochondrial', 'pct_counts_mitochondrial', 'pct_counts_in_top_50_features_mitochondrial', 'pct_counts_in_top_100_features_mitochondrial', 'pct_counts_in_top_200_features_mitochondrial', 'pct_counts_in_top_500_features_mitochondrial', 'total_features_by_counts_ribosomal', 'log10_total_features_by_counts_ribosomal', 'total_counts_ribosomal', 'log10_total_counts_ribosomal', 'pct_counts_ribosomal', 'pct_counts_in_top_50_features_ribosomal', 'pct_counts_in_top_100_features_ribosomal', 'pct_counts_in_top_200_features_ribosomal', 'pct_counts_in_top_500_features_ribosomal', 'size_factor', 'cellassign_cluster_broad', 'cellassign_cluster_specific', 'B.cells..broad.', 'T.cells..broad.', 'other..broad.', 'B.cells', 'Cytotoxic.T.cells', 'CD4.T.cells', 'Tfh', 'other', 'celltype', 'malignant_status_manual', 'celltype_full', 'G1', 'S', 'G2M', 'Cell_Cycle', 't_seurat_cluster', 't_seurat_0.8_cluster', 't_phenograph_cluster', 't_cluster', 'malignant_seurat_cluster', 'malignant_seurat_0.8_cluster', 'malignant_phenograph_cluster', 'malignant_cluster', 'b_seurat_cluster', 'b_seurat_0.8_cluster', 'b_phenograph_cluster', 'b_cluster', 'all_seurat_cluster', 'all_seurat_0.8_cluster', 'all_seurat_1.2_cluster', 'all_sc3_cluster', 'all_SC3_cluster', 'all_cluster', 'all_subset_seurat_cluster', 'all_subset_seurat_0.8_cluster', 'all_subset_seurat_1.2_cluster', 'all_subset_cluster'
var: 'ID', 'is_feature_control', 'is_feature_control_mitochondrial', 'is_feature_control_ribosomal', 'mean_counts', 'log10_mean_counts', 'n_cells_by_counts', 'pct_dropout_by_counts', 'total_counts', 'log10_total_counts'
uns: 'log.exprs.offset'
obsm: 'X_pca', 'X_tsne', 'X_umap'
layers: 'logcounts'
Create and fit CellAssign model#
The anndata object and cell type marker matrix should contain the same genes, so we index into adata to include only the genes from marker_gene_mat.
follicular_bdata = follicular_adata[:, fl_celltype_markers.index].copy()
Then we setup anndata and initialize a CellAssign model. Here we set the size_factor_key to “size_factor”, which is a column in bdata.obs.
Note
A size factor may be defined manually as scaled library size (total UMI count) and should not be placed on the log scale, as the model will do this manually. The library size should be computed before any gene subsetting (in this case, technically, a few notebook cells up).
This can be acheived as follows:
lib_size = adata.X.sum(1)
adata.obs["size_factor"] = lib_size / np.mean(lib_size)
scvi.external.CellAssign.setup_anndata(follicular_bdata, size_factor_key="size_factor")
follicular_model = CellAssign(follicular_bdata, fl_celltype_markers)
follicular_model.train()
Monitored metric elbo_validation did not improve in the last 15 records. Best score: 20.184. Signaling Trainer to stop.
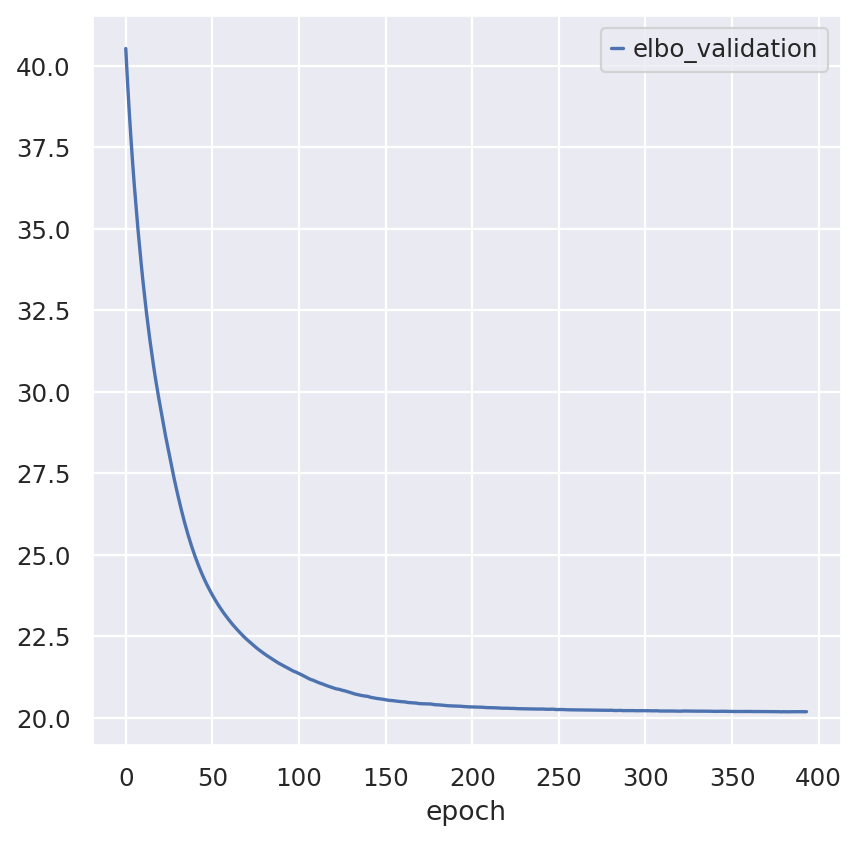
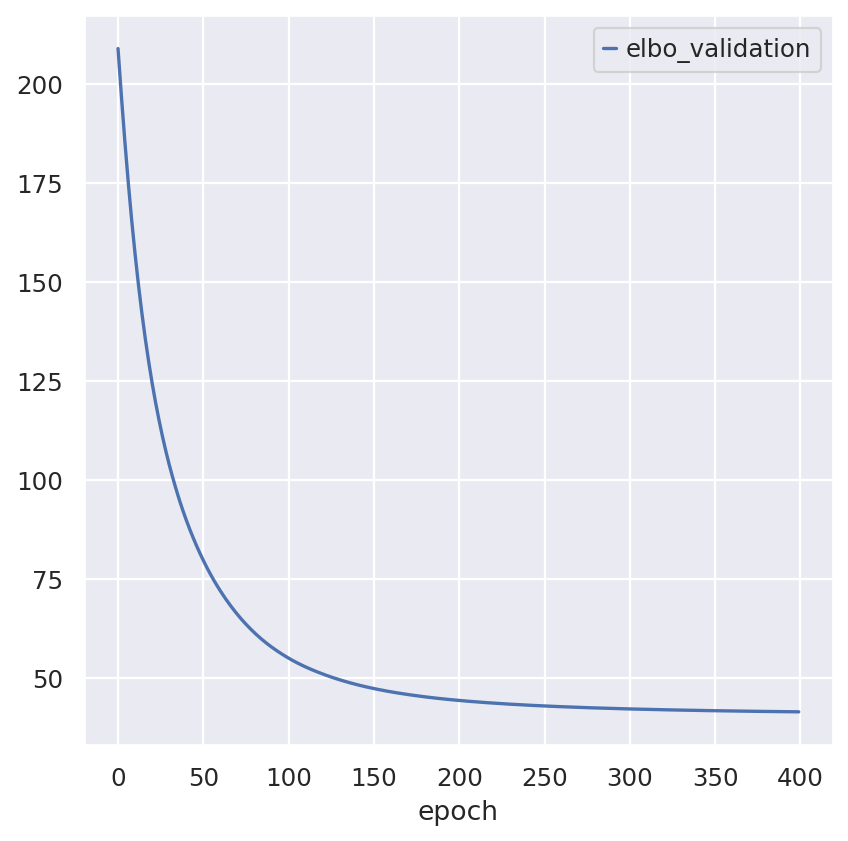
Inspecting the convergence:
follicular_model.history["elbo_validation"].plot()
<Axes: xlabel='epoch'>
Predict and plot assigned cell types#
Predict the soft cell type assignment probability for each cell.
predictions = follicular_model.predict()
predictions.head()
| B cells | Cytotoxic T cells | CD4 T cells | Tfh | other | |
|---|---|---|---|---|---|
| 0 | 1.000000e+00 | 1.971763e-16 | 2.332297e-11 | 4.605773e-13 | 1.090822e-10 |
| 1 | 1.000000e+00 | 1.126303e-18 | 7.889835e-14 | 9.161441e-16 | 5.378042e-13 |
| 2 | 1.000000e+00 | 1.445613e-23 | 1.004261e-18 | 8.941618e-21 | 9.393619e-18 |
| 3 | 1.000000e+00 | 2.512004e-39 | 1.676703e-32 | 2.820753e-35 | 4.558854e-29 |
| 4 | 2.223950e-17 | 2.720671e-13 | 9.994089e-01 | 5.918650e-04 | 9.924171e-19 |
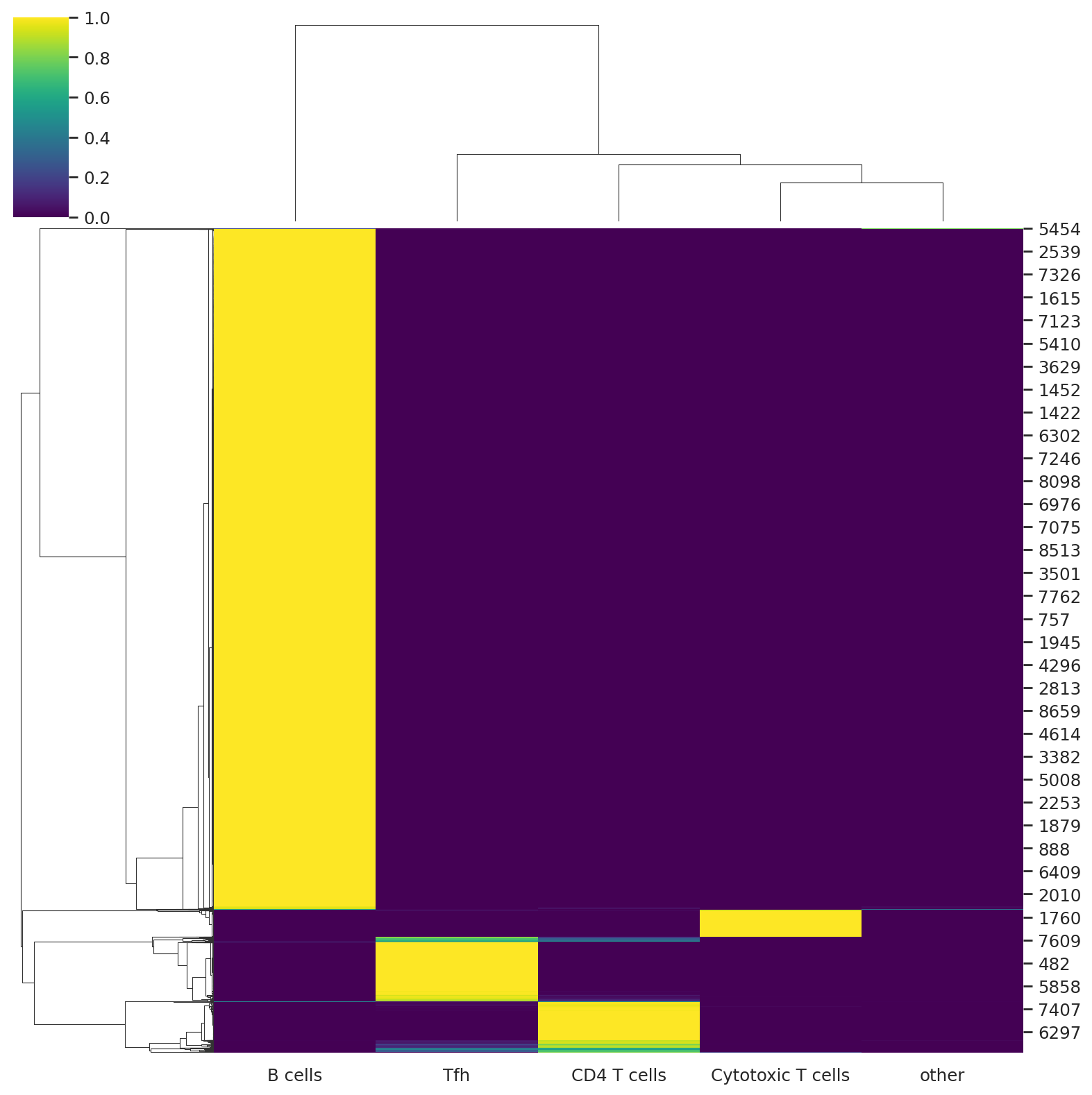
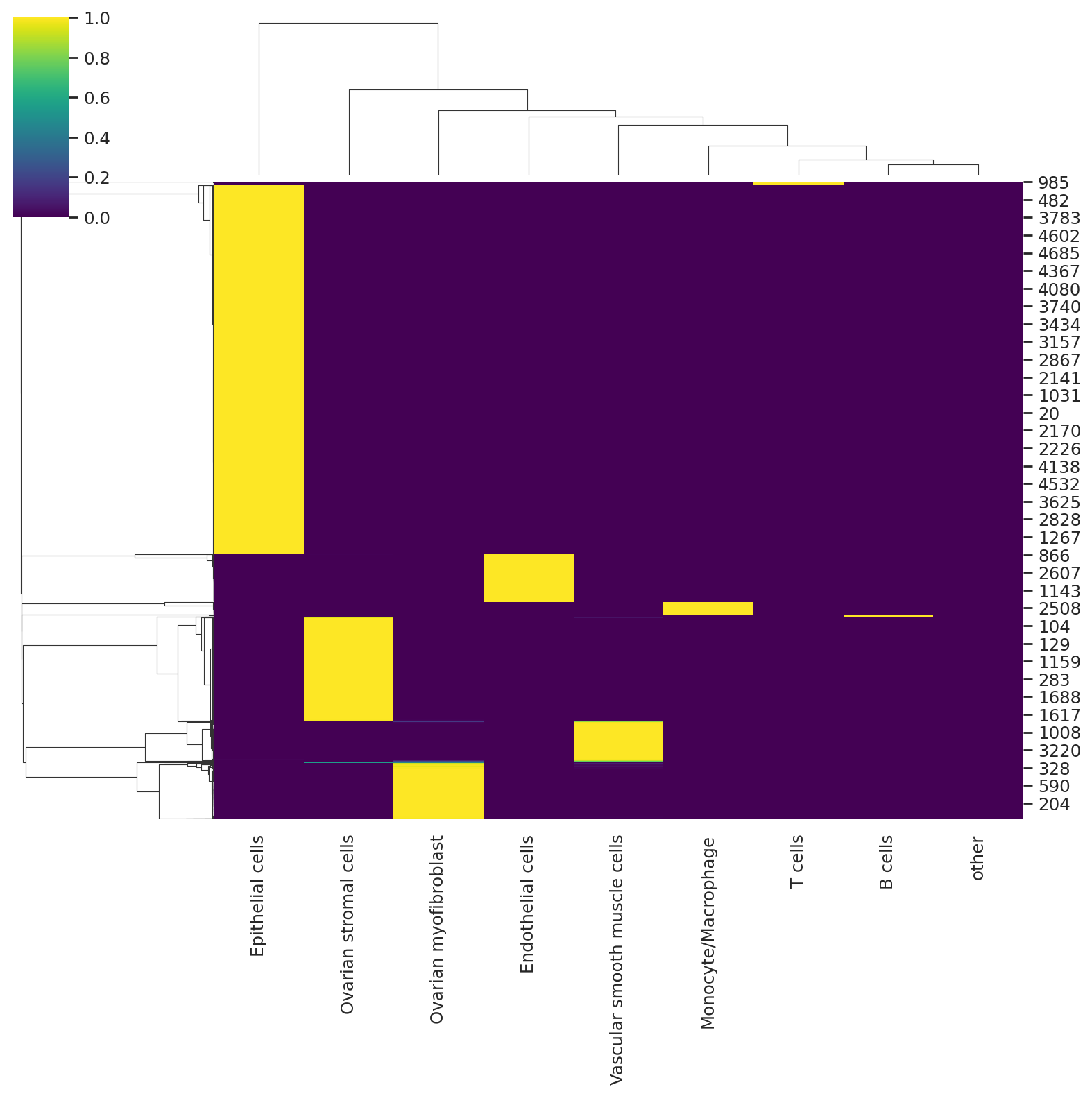
We can visualize the probabilities of assignment with a heatmap that returns the probability matrix for each cell and cell type.
sns.clustermap(predictions, cmap="viridis")
<seaborn.matrix.ClusterGrid at 0x729b84ab1340>
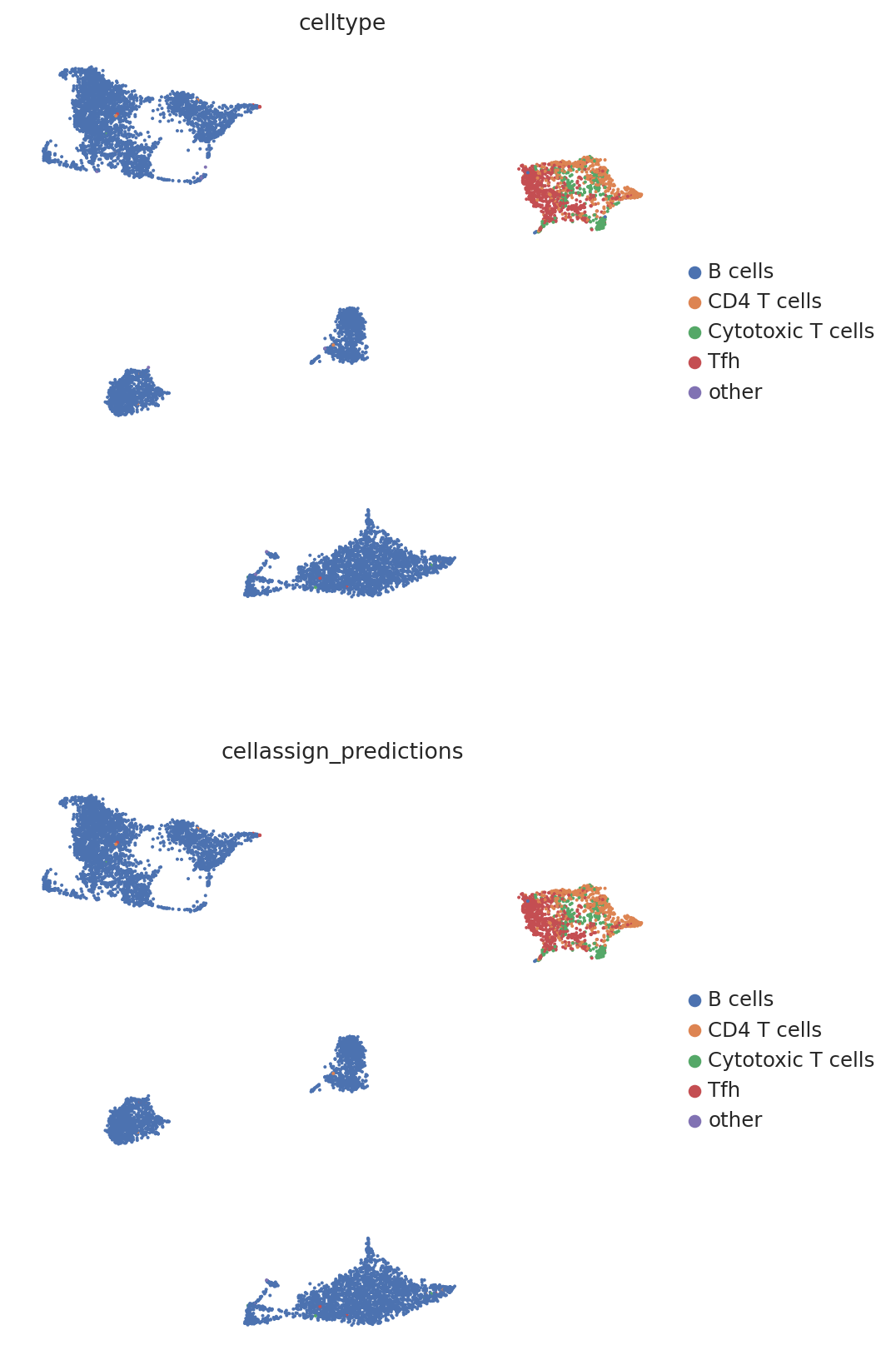
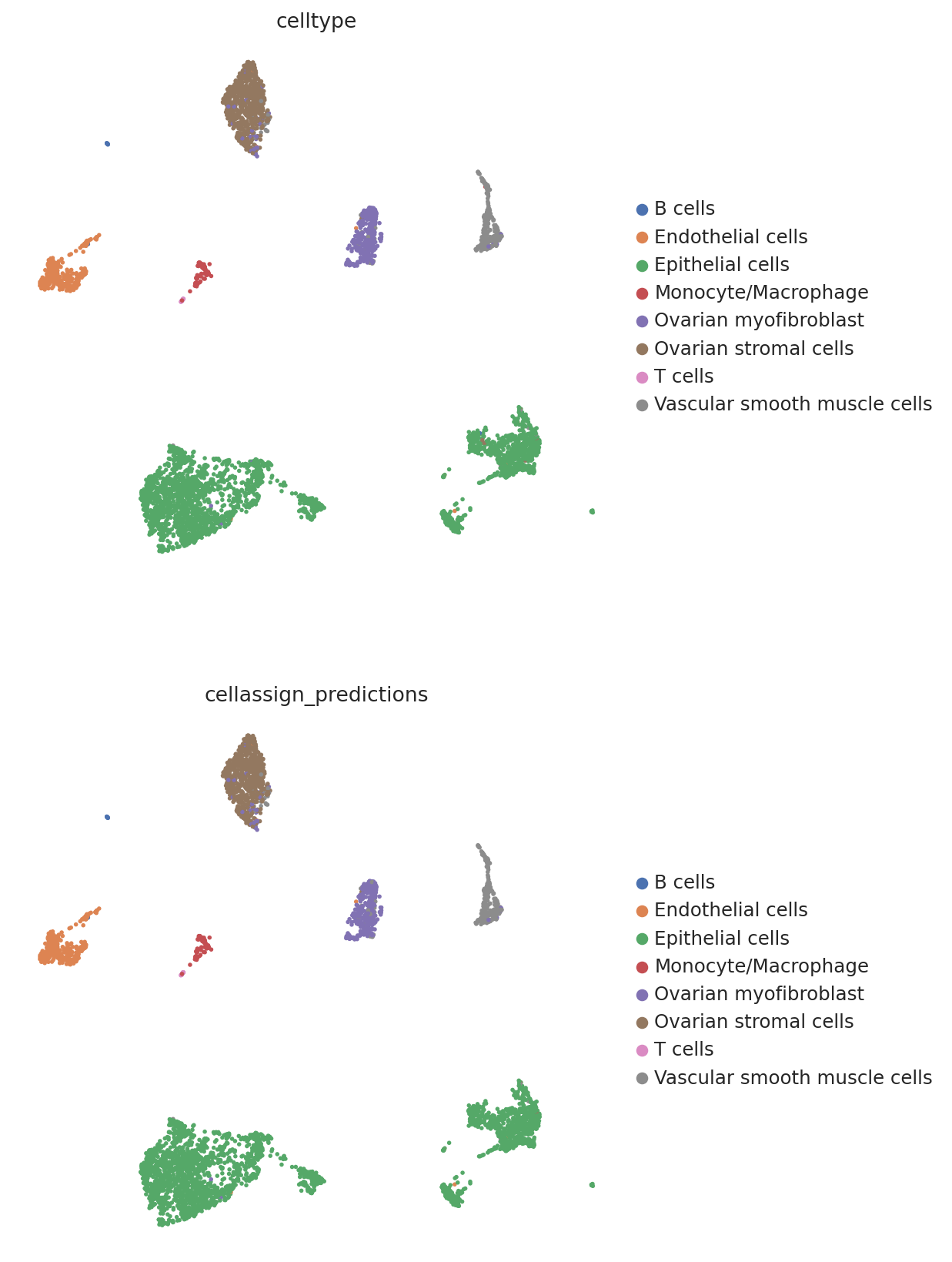
We then create a UMAP plot labeled by maximum probability assignments from the CellAssign model. The left plot contains the true cell types and the right plot contains our model’s predictions.
follicular_bdata.obs["cellassign_predictions"] = predictions.idxmax(axis=1).values
# celltype is the original CellAssign prediction
sc.pl.umap(
follicular_bdata,
color=["celltype", "cellassign_predictions"],
frameon=False,
ncols=1,
)
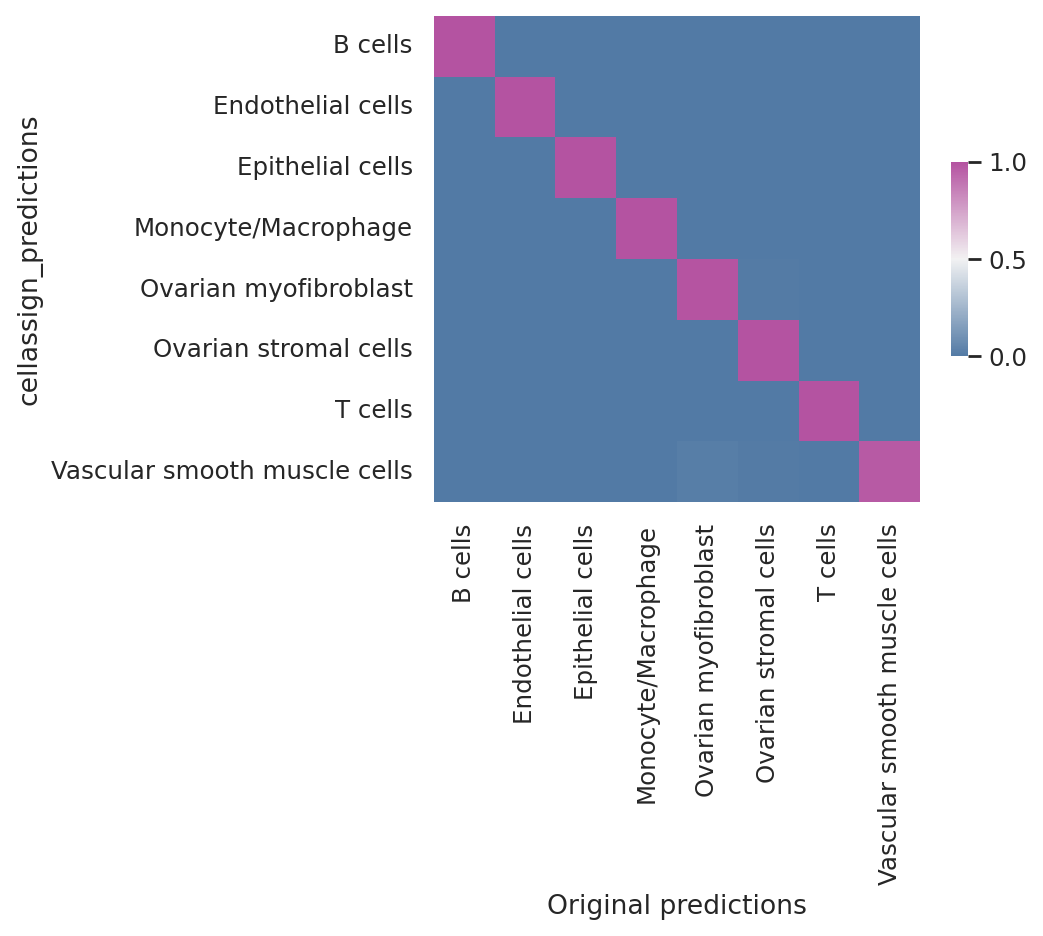
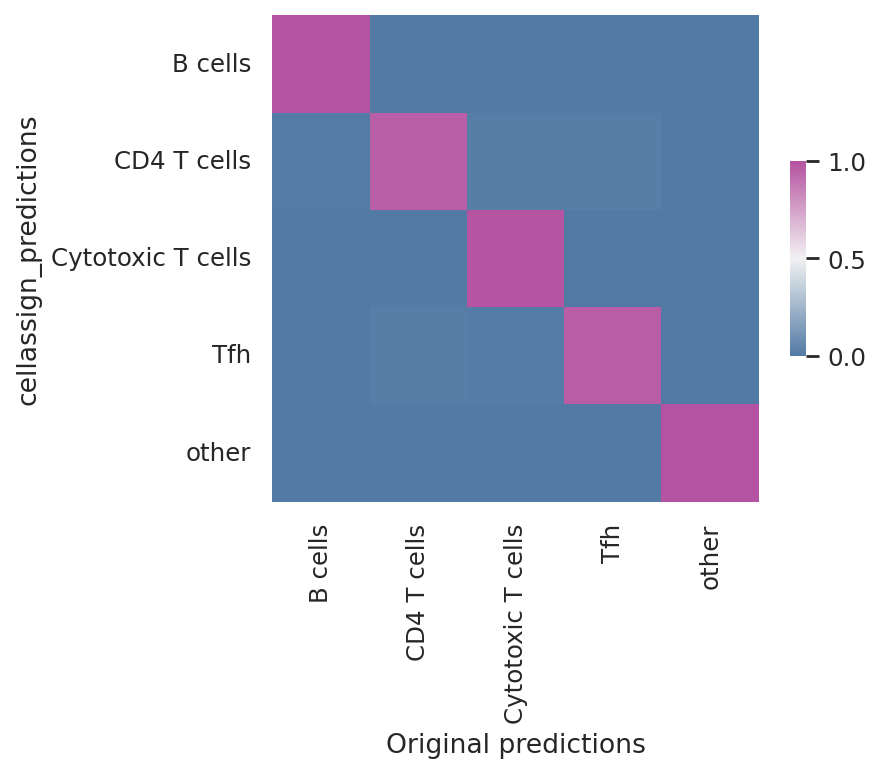
Model reproducibility#
We see that the scvi-tools implementation highly reproduces the original implementation’s predictions.
df = follicular_bdata.obs
confusion_matrix = pd.crosstab(
df["cellassign_predictions"],
df["celltype"],
rownames=["cellassign_predictions"],
colnames=["Original predictions"],
)
confusion_matrix /= confusion_matrix.sum(1).ravel().reshape(-1, 1)
fig, ax = plt.subplots(figsize=(5, 4))
sns.heatmap(
confusion_matrix,
cmap=sns.diverging_palette(245, 320, s=60, as_cmap=True),
ax=ax,
square=True,
cbar_kws={"shrink": 0.4, "aspect": 12},
)
<Axes: xlabel='Original predictions', ylabel='cellassign_predictions'>
HGSC Data#
We can repeat the same process for HGSC data.
hgsc_adata = scvi.data.read_h5ad(sce_hgsc_path)
hgsc_celltype_markers = pd.read_csv(hgsc_celltype_path, index_col=0)
hgsc_adata.var_names_make_unique()
hgsc_adata.obs_names_make_unique()
hgsc_adata
AnnData object with n_obs × n_vars = 4848 × 33694
obs: 'Sample', 'dataset', 'patient', 'timepoint', 'site', 'sample_barcode', 'is_cell_control', 'total_features_by_counts', 'log10_total_features_by_counts', 'total_counts', 'log10_total_counts', 'pct_counts_in_top_50_features', 'pct_counts_in_top_100_features', 'pct_counts_in_top_200_features', 'pct_counts_in_top_500_features', 'total_features_by_counts_endogenous', 'log10_total_features_by_counts_endogenous', 'total_counts_endogenous', 'log10_total_counts_endogenous', 'pct_counts_endogenous', 'pct_counts_in_top_50_features_endogenous', 'pct_counts_in_top_100_features_endogenous', 'pct_counts_in_top_200_features_endogenous', 'pct_counts_in_top_500_features_endogenous', 'total_features_by_counts_feature_control', 'log10_total_features_by_counts_feature_control', 'total_counts_feature_control', 'log10_total_counts_feature_control', 'pct_counts_feature_control', 'pct_counts_in_top_50_features_feature_control', 'pct_counts_in_top_100_features_feature_control', 'pct_counts_in_top_200_features_feature_control', 'pct_counts_in_top_500_features_feature_control', 'total_features_by_counts_mitochondrial', 'log10_total_features_by_counts_mitochondrial', 'total_counts_mitochondrial', 'log10_total_counts_mitochondrial', 'pct_counts_mitochondrial', 'pct_counts_in_top_50_features_mitochondrial', 'pct_counts_in_top_100_features_mitochondrial', 'pct_counts_in_top_200_features_mitochondrial', 'pct_counts_in_top_500_features_mitochondrial', 'total_features_by_counts_ribosomal', 'log10_total_features_by_counts_ribosomal', 'total_counts_ribosomal', 'log10_total_counts_ribosomal', 'pct_counts_ribosomal', 'pct_counts_in_top_50_features_ribosomal', 'pct_counts_in_top_100_features_ribosomal', 'pct_counts_in_top_200_features_ribosomal', 'pct_counts_in_top_500_features_ribosomal', 'size_factor', 'cellassign_cluster_broad', 'cellassign_cluster_specific', 'B.cells..broad.', 'T.cells..broad.', 'Monocyte.Macrophage..broad.', 'Epithelial.cells..broad.', 'Ovarian.stromal.cells..broad.', 'Ovarian.myofibroblast..broad.', 'Vascular.smooth.muscle.cells..broad.', 'Endothelial.cells..broad.', 'other..broad.', 'B.cells', 'CD4.T.cells', 'Cytotoxic.T.cells', 'Monocyte.Macrophage', 'Epithelial.cells', 'Ovarian.stromal.cells', 'Ovarian.myofibroblast', 'Vascular.smooth.muscle.cells', 'Endothelial.cells', 'other', 'celltype', 'G1', 'S', 'G2M', 'Cell_Cycle', 'epithelial_seurat_cluster', 'epithelial_seurat_0.2_cluster', 'epithelial_phenograph_cluster', 'epithelial_sc3_cluster', 'epithelial_SC3_cluster', 'epithelial_cluster', 'all_seurat_cluster', 'all_seurat_0.8_cluster', 'all_seurat_1.2_cluster', 'all_sc3_cluster', 'all_SC3_cluster', 'all_cluster', 'all_subset_seurat_cluster', 'all_subset_seurat_0.8_cluster', 'all_subset_seurat_1.2_cluster', 'all_subset_cluster'
var: 'ID', 'is_feature_control', 'is_feature_control_mitochondrial', 'is_feature_control_ribosomal', 'mean_counts', 'log10_mean_counts', 'n_cells_by_counts', 'pct_dropout_by_counts', 'total_counts', 'log10_total_counts'
uns: 'log.exprs.offset'
obsm: 'X_pca', 'X_tsne', 'X_umap'
layers: 'logcounts'
Create and fit CellAssign model#
hgsc_bdata = hgsc_adata[:, hgsc_celltype_markers.index].copy()
scvi.external.CellAssign.setup_anndata(hgsc_bdata, "size_factor")
hgsc_model = CellAssign(hgsc_bdata, hgsc_celltype_markers)
hgsc_model.train()
hgsc_model.history["elbo_validation"].plot()
<Axes: xlabel='epoch'>
Predict and plot assigned cell types#
predictions_hgsc = hgsc_model.predict()
predictions.head()
| B cells | Cytotoxic T cells | CD4 T cells | Tfh | other | |
|---|---|---|---|---|---|
| 0 | 1.000000e+00 | 1.971763e-16 | 2.332297e-11 | 4.605773e-13 | 1.090822e-10 |
| 1 | 1.000000e+00 | 1.126303e-18 | 7.889835e-14 | 9.161441e-16 | 5.378042e-13 |
| 2 | 1.000000e+00 | 1.445613e-23 | 1.004261e-18 | 8.941618e-21 | 9.393619e-18 |
| 3 | 1.000000e+00 | 2.512004e-39 | 1.676703e-32 | 2.820753e-35 | 4.558854e-29 |
| 4 | 2.223950e-17 | 2.720671e-13 | 9.994089e-01 | 5.918650e-04 | 9.924171e-19 |
sns.clustermap(predictions_hgsc, cmap="viridis")
<seaborn.matrix.ClusterGrid at 0x729b1835ab70>
hgsc_bdata.obs["cellassign_predictions"] = predictions_hgsc.idxmax(axis=1).values
sc.pl.umap(
hgsc_bdata,
color=["celltype", "cellassign_predictions"],
ncols=1,
frameon=False,
)
Model reproducibility#
df = hgsc_bdata.obs
confusion_matrix = pd.crosstab(
df["cellassign_predictions"],
df["celltype"],
rownames=["cellassign_predictions"],
colnames=["Original predictions"],
)
confusion_matrix /= confusion_matrix.sum(1).ravel().reshape(-1, 1)
fig, ax = plt.subplots(figsize=(5, 4))
sns.heatmap(
confusion_matrix,
cmap=sns.diverging_palette(245, 320, s=60, as_cmap=True),
ax=ax,
square=True,
cbar_kws={"shrink": 0.4, "aspect": 12},
)
<Axes: xlabel='Original predictions', ylabel='cellassign_predictions'>