ScBasset: Analyzing scATACseq data#
Warning
SCBASSET’s development is still in progress. The current version may not fully reproduce the original implementation’s results.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import tempfile
import matplotlib.pyplot as plt
import muon
import numpy as np
import scanpy as sc
import scvi
import seaborn as sns
import torch
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.2
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Loading data and preprocessing#
Throughout this tutorial, we use sample multiome data from 10X of 10K PBMCs.
url = "https://cf.10xgenomics.com/samples/cell-arc/2.0.0/10k_PBMC_Multiome_nextgem_Chromium_X/10k_PBMC_Multiome_nextgem_Chromium_X_filtered_feature_bc_matrix.h5"
mdata = muon.read_10x_h5("data/multiome10k.h5mu", backup_url=url)
Added `interval` annotation for features from data/multiome10k.h5mu
mdata
MuData object with n_obs × n_vars = 10970 × 148344
var: 'gene_ids', 'feature_types', 'genome', 'interval'
2 modalities
rna: 10970 x 36601
var: 'gene_ids', 'feature_types', 'genome', 'interval'
atac: 10970 x 111743
var: 'gene_ids', 'feature_types', 'genome', 'interval'adata = mdata.mod["atac"]
We can use scanpy functions to handle, filter, and manipulate the data. In our case, we might want to filter out peaks that are rarely detected, to make the model train faster:
print(adata.shape)
# compute the threshold: 5% of the cells
min_cells = int(adata.shape[0] * 0.05)
# in-place filtering of regions
sc.pp.filter_genes(adata, min_cells=min_cells)
print(adata.shape)
(10970, 111743)
(10970, 37054)
adata.var
| gene_ids | feature_types | genome | interval | n_cells | |
|---|---|---|---|---|---|
| chr1:629395-630394 | chr1:629395-630394 | Peaks | GRCh38 | chr1:629395-630394 | 1422 |
| chr1:633578-634591 | chr1:633578-634591 | Peaks | GRCh38 | chr1:633578-634591 | 4536 |
| chr1:778283-779200 | chr1:778283-779200 | Peaks | GRCh38 | chr1:778283-779200 | 5981 |
| chr1:816873-817775 | chr1:816873-817775 | Peaks | GRCh38 | chr1:816873-817775 | 564 |
| chr1:827067-827949 | chr1:827067-827949 | Peaks | GRCh38 | chr1:827067-827949 | 3150 |
| ... | ... | ... | ... | ... | ... |
| GL000219.1:44739-45583 | GL000219.1:44739-45583 | Peaks | GRCh38 | GL000219.1:44739-45583 | 781 |
| GL000219.1:45726-46446 | GL000219.1:45726-46446 | Peaks | GRCh38 | GL000219.1:45726-46446 | 639 |
| GL000219.1:99267-100169 | GL000219.1:99267-100169 | Peaks | GRCh38 | GL000219.1:99267-100169 | 6830 |
| KI270726.1:41483-42332 | KI270726.1:41483-42332 | Peaks | GRCh38 | KI270726.1:41483-42332 | 605 |
| KI270713.1:21453-22374 | KI270713.1:21453-22374 | Peaks | GRCh38 | KI270713.1:21453-22374 | 6247 |
37054 rows × 5 columns
split_interval = adata.var["gene_ids"].str.split(":", expand=True)
adata.var["chr"] = split_interval[0]
split_start_end = split_interval[1].str.split("-", expand=True)
adata.var["start"] = split_start_end[0].astype(int)
adata.var["end"] = split_start_end[1].astype(int)
adata.var
| gene_ids | feature_types | genome | interval | n_cells | chr | start | end | |
|---|---|---|---|---|---|---|---|---|
| chr1:629395-630394 | chr1:629395-630394 | Peaks | GRCh38 | chr1:629395-630394 | 1422 | chr1 | 629395 | 630394 |
| chr1:633578-634591 | chr1:633578-634591 | Peaks | GRCh38 | chr1:633578-634591 | 4536 | chr1 | 633578 | 634591 |
| chr1:778283-779200 | chr1:778283-779200 | Peaks | GRCh38 | chr1:778283-779200 | 5981 | chr1 | 778283 | 779200 |
| chr1:816873-817775 | chr1:816873-817775 | Peaks | GRCh38 | chr1:816873-817775 | 564 | chr1 | 816873 | 817775 |
| chr1:827067-827949 | chr1:827067-827949 | Peaks | GRCh38 | chr1:827067-827949 | 3150 | chr1 | 827067 | 827949 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| GL000219.1:44739-45583 | GL000219.1:44739-45583 | Peaks | GRCh38 | GL000219.1:44739-45583 | 781 | GL000219.1 | 44739 | 45583 |
| GL000219.1:45726-46446 | GL000219.1:45726-46446 | Peaks | GRCh38 | GL000219.1:45726-46446 | 639 | GL000219.1 | 45726 | 46446 |
| GL000219.1:99267-100169 | GL000219.1:99267-100169 | Peaks | GRCh38 | GL000219.1:99267-100169 | 6830 | GL000219.1 | 99267 | 100169 |
| KI270726.1:41483-42332 | KI270726.1:41483-42332 | Peaks | GRCh38 | KI270726.1:41483-42332 | 605 | KI270726.1 | 41483 | 42332 |
| KI270713.1:21453-22374 | KI270713.1:21453-22374 | Peaks | GRCh38 | KI270713.1:21453-22374 | 6247 | KI270713.1 | 21453 | 22374 |
37054 rows × 8 columns
# Filter out non-chromosomal regions
mask = adata.var["chr"].str.startswith("chr")
adata = adata[:, mask].copy()
scvi.data.add_dna_sequence(
adata,
genome_name="GRCh38",
genome_dir="data",
chr_var_key="chr",
start_var_key="start",
end_var_key="end",
)
adata
Working...: 100%|██████████| 24/24 [00:02<00:00, 10.90it/s]
AnnData object with n_obs × n_vars = 10970 × 37042
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'n_cells', 'chr', 'start', 'end'
varm: 'dna_sequence', 'dna_code'
adata.varm["dna_sequence"]
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ... | 1334 | 1335 | 1336 | 1337 | 1338 | 1339 | 1340 | 1341 | 1342 | 1343 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr1:629395-630394 | C | A | C | T | C | T | C | C | C | C | ... | C | T | A | T | A | T | C | T | A | A |
| chr1:633578-634591 | G | A | A | A | T | A | G | G | G | C | ... | T | A | A | A | T | C | C | C | C | T |
| chr1:778283-779200 | C | G | C | C | C | G | G | C | T | A | ... | G | A | C | A | G | G | A | G | T | T |
| chr1:816873-817775 | A | A | T | T | C | A | T | A | T | G | ... | T | T | A | G | C | G | G | C | T | G |
| chr1:827067-827949 | C | T | C | T | C | C | T | G | C | C | ... | C | G | T | T | A | T | T | A | A | T |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| chrY:19077075-19078016 | A | C | G | A | C | C | T | C | C | C | ... | C | A | T | A | G | T | T | C | T | A |
| chrY:19567013-19567787 | G | G | A | G | T | C | T | G | G | G | ... | T | C | T | C | T | T | C | G | T | T |
| chrY:19744368-19745303 | T | A | T | T | T | T | T | G | T | C | ... | A | T | G | T | G | G | A | A | A | T |
| chrY:20575244-20576162 | T | T | T | A | C | T | G | T | C | T | ... | G | A | G | T | G | T | A | A | C | A |
| chrY:21240021-21240909 | A | T | T | G | G | C | C | C | C | C | ... | C | C | T | T | T | C | T | G | A | G |
37042 rows × 1344 columns
Creating and training the model#
We can now set up the AnnData object, which will ensure everything the model needs is in place for training.
This is also the stage where we can condition the model on additional covariates, which encourages the model to remove the impact of those covariates from the learned latent space. Our sample data is a single batch, so we won’t demonstrate this directly, but it can be done simply by setting the batch_key argument to the annotation to be used as a batch covariate (must be a valid key in adata.obs) .
# alternatively load the local preprocessed data
# import os
# temp_dir_obj = tempfile.TemporaryDirectory()
# adata_path = os.path.join(temp_dir_obj.name, "adata_scbasset.h5ad")
# adata = sc.read(adata_path, backup_url="https://exampledata.scverse.org/scvi-tools/adata_scbasset.h5ad")
# adata
AnnData object with n_obs × n_vars = 10970 × 37042
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'n_cells', 'chr', 'start', 'end'
varm: 'dna_code', 'dna_sequence'
bdata = adata.transpose()
bdata.layers["binary"] = (bdata.X.copy() > 0).astype(float)
scvi.external.SCBASSET.setup_anndata(bdata, layer="binary", dna_code_key="dna_code")
INFO Using column names from columns of adata.obsm['dna_code']
We can now create a scBasset model object and train it!
Note
The default max epochs is set to 1000, but in practice scBasset stops early once the model converges, which especially for large datasets (which require fewer epochs to converge, since each epoch includes letting the model view more data).
Here we are using 16 bit precision which uses less memory without sacrificing performance.
bas = scvi.external.SCBASSET(bdata)
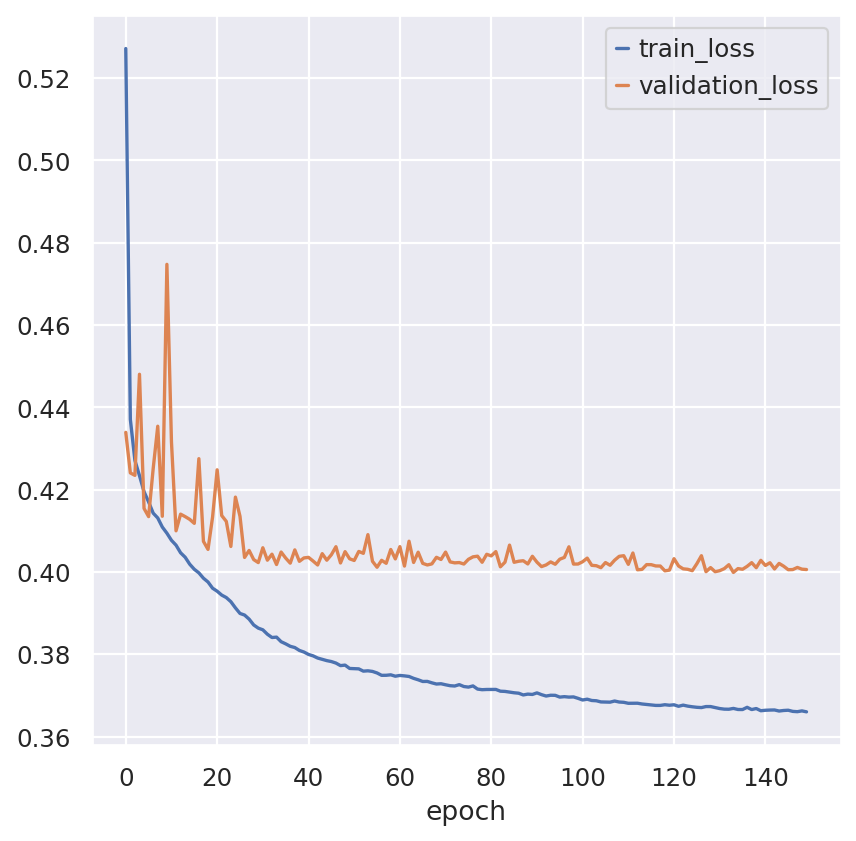
bas.train(max_epochs=150, precision=16)
fig, ax = plt.subplots()
bas.history_["train_loss"].plot(ax=ax)
bas.history_["validation_loss"].plot(ax=ax)
<Axes: xlabel='epoch'>
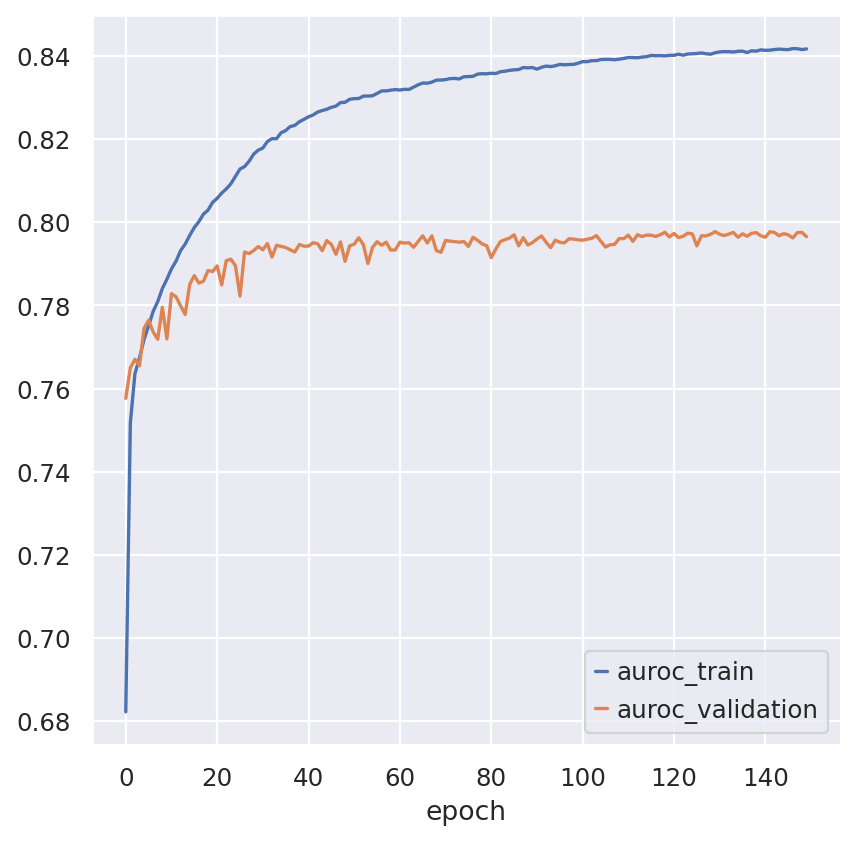
fig, ax = plt.subplots()
bas.history_["auroc_train"].plot(ax=ax)
bas.history_["auroc_validation"].plot(ax=ax)
<Axes: xlabel='epoch'>
Visualizing and analyzing the latent space#
We can now use the trained model to visualize, cluster, and analyze the data. We first extract the latent representation from the model, and save it back into our AnnData object:
latent = bas.get_latent_representation()
adata.obsm["X_scbasset"] = latent
print(latent.shape)
(10970, 32)
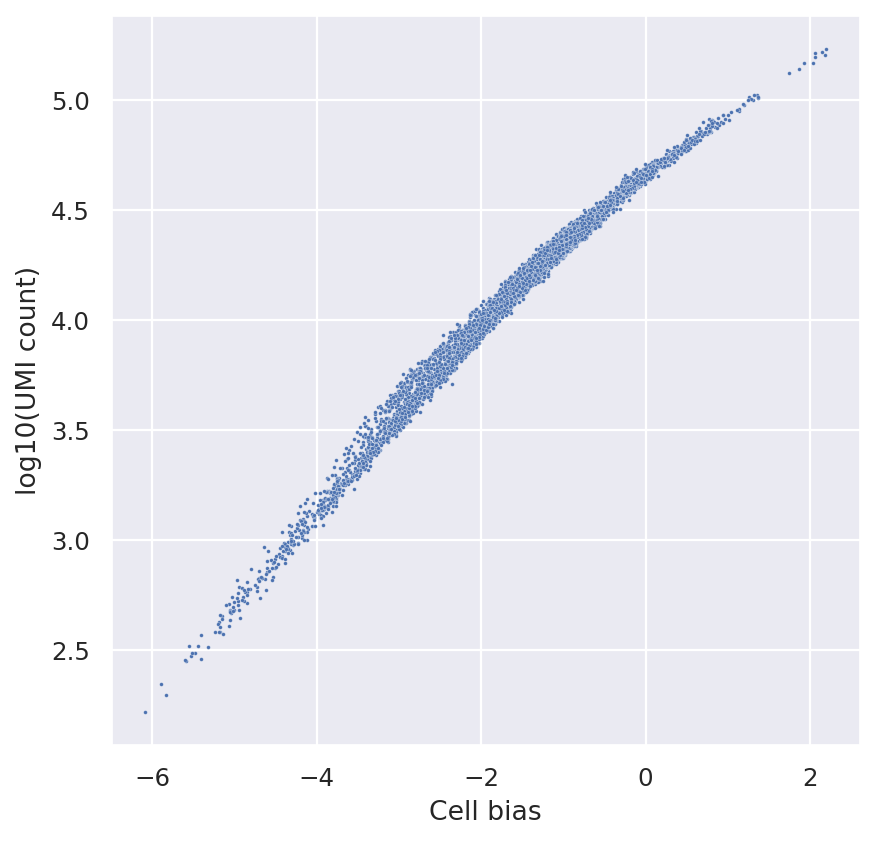
sns.scatterplot(
x=bas.get_cell_bias(),
y=np.log10(np.asarray(adata.X.sum(1))).ravel(),
s=3,
)
plt.xlabel("Cell bias")
plt.ylabel("log10(UMI count)")
Text(0, 0.5, 'log10(UMI count)')
We can now use scanpy functions to cluster and visualize our latent space:
# compute the k-nearest-neighbor graph that is used in both clustering and umap algorithms
sc.pp.neighbors(adata, use_rep="X_scbasset")
# compute the umap
sc.tl.umap(adata)
sc.tl.leiden(adata, key_added="leiden_scbasset")
sc.pl.umap(adata, color="leiden_scbasset")

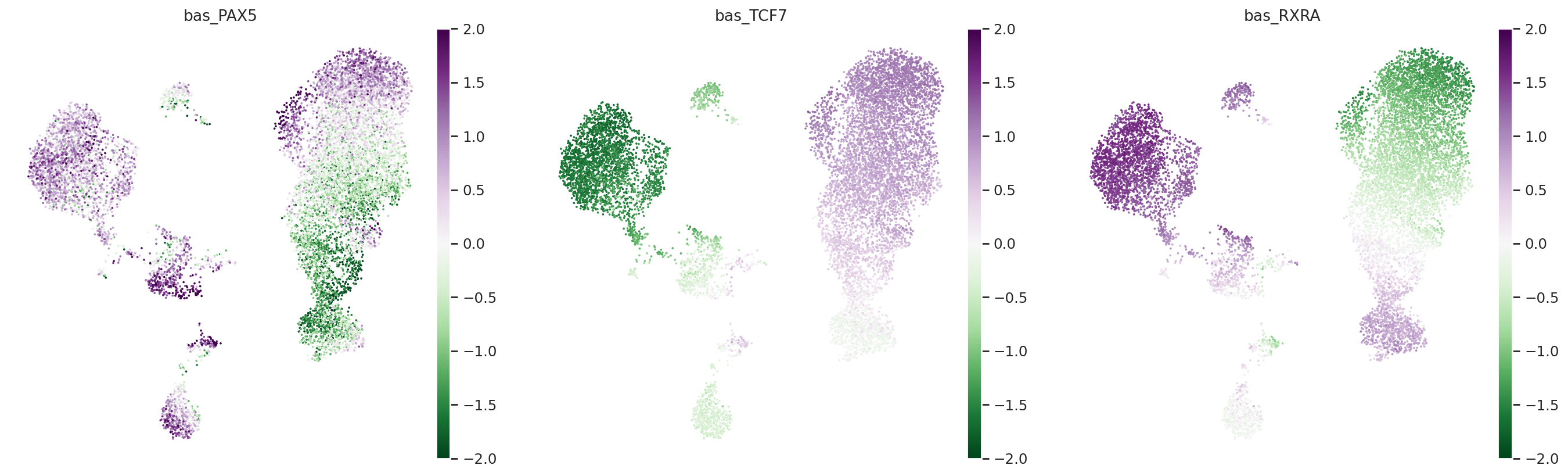
Score TF activity#
We will now use the motif injection procedure to infer the activity of human transcription factors using their motifs.
This process involves downloading a library of (1) random dinucleotide shuffled sequences and (2) random sequences with a known motif injected. We infer the accessibility of all the random sequences and all the motif injected sequences in every cell using the SCBASSET model. We then compute the difference in activity for the motif injected sequences and the random sequences. This difference serves as an estimate for the likelihood that a given motif is accessible in each cell, and therefore an estimate of a corresponding transcription factor’s activity.
Any library with sequences of the appropriate size can be used. By default, we provide the human TF motif library used in the scBasset paper. The library is downloaded to a local folder (default: ./scbasset_motifs). Each motif is stored in a specific FASTA file in the {library_path}/shuffled_peaks_motifs subdirectory. To see all available motifs, simply glob the path (e.g. Path("./scbasset_motifs/shuffled_peaks_motifs").glob("*.fasta")).
tfs = ["PAX5", "TCF7", "RXRA"]
for tf in tfs:
adata.obs[f"bas_{tf}"] = bas.get_tf_activity(
tf=tf,
motif_dir="data/motifs",
)
INFO Downloading motif set to: data/motifs
INFO Downloading file at data/motifs/human_motifs.tar.gz
INFO Download and extraction complete.
sc.pl.umap(
adata,
color=[f"bas_{tf}" for tf in tfs],
cmap="PRGn_r",
vmin=-2,
vmax=2,
)