Reference mapping with scvi-tools#
This tutorial covers the usage of the scArches method with SCVI, SCANVI, and TOTALVI.
This particular workflow is useful in the case where a model is trained on some data (called reference here) and new samples are received (called query). The goal is to analyze these samples in the context of the reference, by mapping the query cells to the same reference latent space. This workflow may also be used in the scarches package, but here we demonstrate using only scvi-tools.
Imports and scvi-tools installation (colab)#
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import os
import tempfile
import anndata
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import scrublet as scr
import scvi
import seaborn as sns
import torch
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.0.3
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Reference mapping with SCVI#
Here we use the pancreas dataset described in the scIB manuscript, that is also widely used to benchmark integration methods.
pancreas_adata_path = os.path.join(save_dir.name, "pancreas.h5ad")
pancreas_adata = sc.read(
pancreas_adata_path,
backup_url="https://figshare.com/ndownloader/files/24539828",
)
pancreas_adata
AnnData object with n_obs × n_vars = 16382 × 19093
obs: 'tech', 'celltype', 'size_factors'
layers: 'counts'
pancreas_adata.obs["tech"].value_counts()
tech
inDrop3 3605
smartseq2 2394
celseq2 2285
inDrop1 1937
inDrop2 1724
smarter 1492
inDrop4 1303
celseq 1004
fluidigmc1 638
Name: count, dtype: int64
We consider the SS2 and CelSeq2 samples as query, and all the others as reference.
query_mask = np.array(
[s in ["smartseq2", "celseq2"] for s in pancreas_adata.obs["tech"]]
)
pancreas_ref = pancreas_adata[~query_mask].copy()
pancreas_query = pancreas_adata[query_mask].copy()
We run highly variable gene selection on the reference data and use these same genes for the query data.
sc.pp.highly_variable_genes(
pancreas_ref, n_top_genes=2000, batch_key="tech", subset=True
)
pancreas_query = pancreas_query[:, pancreas_ref.var_names].copy()
Train reference#
We train the reference using the standard SCVI workflow, except we add a few non-default parameters that were identified to work well with scArches.
scvi.model.SCVI.setup_anndata(pancreas_ref, batch_key="tech", layer="counts")
scvi_ref = scvi.model.SCVI(
pancreas_ref,
use_layer_norm="both",
use_batch_norm="none",
encode_covariates=True,
dropout_rate=0.2,
n_layers=2,
)
scvi_ref.train()
Epoch 1/400: 0%| | 0/400 [00:00<?, ?it/s]Epoch 2/400: 0%| | 1/400 [00:00<06:28, 1.03it/s, v_num=1, train_loss_step=971, train_loss_epoch=1.3e+3]Epoch 400/400: 100%|██████████| 400/400 [03:10<00:00, 2.08it/s, v_num=1, train_loss_step=696, train_loss_epoch=787] Epoch 400/400: 100%|██████████| 400/400 [03:10<00:00, 2.10it/s, v_num=1, train_loss_step=696, train_loss_epoch=787]
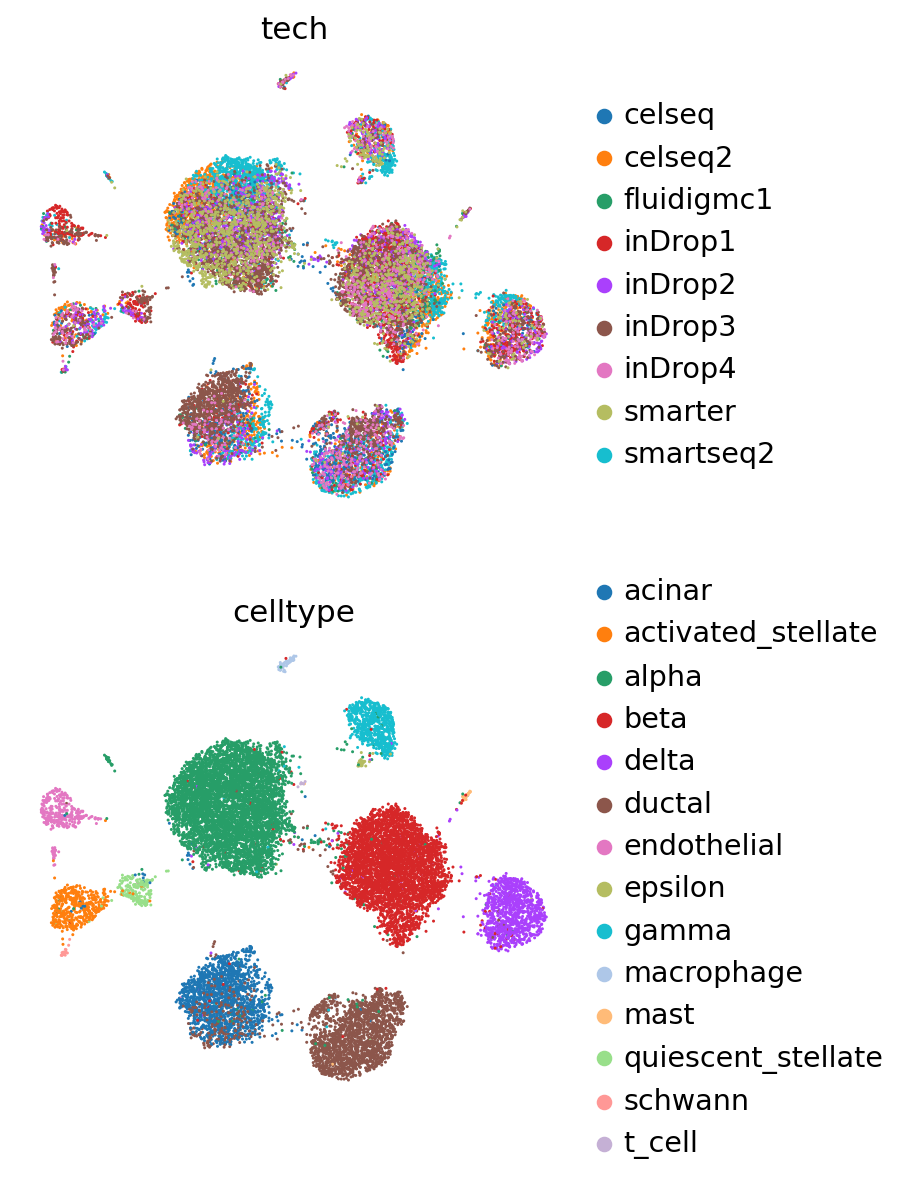
Now we obtain the latent representation, and use Scanpy to visualize with UMAP.
SCVI_LATENT_KEY = "X_scVI"
pancreas_ref.obsm[SCVI_LATENT_KEY] = scvi_ref.get_latent_representation()
sc.pp.neighbors(pancreas_ref, use_rep=SCVI_LATENT_KEY)
sc.tl.leiden(pancreas_ref)
sc.tl.umap(pancreas_ref)
sc.pl.umap(
pancreas_ref,
color=["tech", "celltype"],
frameon=False,
ncols=1,
)

Update with query#
We can load a new model with the query data either using
The saved reference model
The instance of the reference model
scvi_ref_path = os.path.join(save_dir.name, "pancreas_scvi_ref")
scvi_ref.save(scvi_ref_path, overwrite=True)
First we validate that our query data is ready to be loaded into the reference model. Here we run prepare_query_anndata, which reorders the genes and pads any missing genes with 0s. This should generally be run before reference mapping with scArches to ensure data correctness. In the case of this tutorial, nothing happens as the query data is already “correct”.
# both are valid
scvi.model.SCVI.prepare_query_anndata(pancreas_query, scvi_ref_path)
scvi.model.SCVI.prepare_query_anndata(pancreas_query, scvi_ref)
INFO File /tmp/tmp2qkhcse5/pancreas_scvi_ref/model.pt already downloaded
INFO Found 100.0% reference vars in query data.
INFO Found 100.0% reference vars in query data.
Now we create the new query model instance.
# both are valid
scvi_query = scvi.model.SCVI.load_query_data(
pancreas_query,
scvi_ref_path,
)
scvi_query = scvi.model.SCVI.load_query_data(
pancreas_query,
scvi_ref,
)
INFO File /tmp/tmp2qkhcse5/pancreas_scvi_ref/model.pt already downloaded
This is a typical SCVI object, and after training, can be used in any defined way.
For training the query data, we recommend using a weight_decay of 0.0. This ensures the latent representation of the reference cells will remain exactly the same if passing them through this new query model.
scvi_query.train(max_epochs=200, plan_kwargs={"weight_decay": 0.0})
pancreas_query.obsm[SCVI_LATENT_KEY] = scvi_query.get_latent_representation()
Epoch 1/200: 0%| | 1/200 [00:00<00:37, 5.25it/s, v_num=1, train_loss_step=1.98e+3, train_loss_epoch=1.94e+3]Epoch 2/200: 0%| | 1/200 [00:00<00:37, 5.25it/s, v_num=1, train_loss_step=1.98e+3, train_loss_epoch=1.94e+3]Epoch 4/200: 2%|▏ | 3/200 [00:00<00:37, 5.26it/s, v_num=1, train_loss_step=1.91e+3, train_loss_epoch=1.88e+3]Epoch 200/200: 100%|██████████| 200/200 [00:37<00:00, 5.32it/s, v_num=1, train_loss_step=1.87e+3, train_loss_epoch=1.75e+3]Epoch 200/200: 100%|██████████| 200/200 [00:37<00:00, 5.31it/s, v_num=1, train_loss_step=1.87e+3, train_loss_epoch=1.75e+3]
sc.pp.neighbors(pancreas_query, use_rep=SCVI_LATENT_KEY)
sc.tl.leiden(pancreas_query)
sc.tl.umap(pancreas_query)
sc.pl.umap(
pancreas_query,
color=["tech", "celltype"],
frameon=False,
ncols=1,
)

Visualize reference and query#
pancreas_full = anndata.concat([pancreas_query, pancreas_ref])
pancreas_full
AnnData object with n_obs × n_vars = 16382 × 2000
obs: 'tech', 'celltype', 'size_factors', '_scvi_batch', '_scvi_labels', 'leiden'
obsm: 'X_scVI', 'X_umap'
layers: 'counts'
The concatenated object has the latent representations of both reference and query, but we are also able to reobtain these values using the query model.
pancreas_full.obsm[SCVI_LATENT_KEY] = scvi_query.get_latent_representation(
pancreas_full
)
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
sc.pp.neighbors(pancreas_full, use_rep=SCVI_LATENT_KEY)
sc.tl.leiden(pancreas_full)
sc.tl.umap(pancreas_full)
sc.pl.umap(
pancreas_full,
color=["tech", "celltype"],
frameon=False,
ncols=1,
)
Reference mapping with SCANVI#
We’ll use the same Pancreas dataset, this time we set it up such that we register that the dataset has labels.
The advantage of SCANVI is that we’ll be able to predict the cell type labels of the query dataset. In the case of SCVI, a separate classifier (e.g., nearest-neighbor, random forest, etc.) would have to be trained on the reference latent space.
Train reference#
SCANVI tends to perform better in situations where it has been initialized using a pre-trained SCVI model. In this case, we will use vae_ref that we have already trained above. In other words, a typical SCANVI workflow will be:
scvi_model = SCVI(adata_ref, **arches_params)
scvi_model.train()
scanvi_model = SCANVI.from_scvi_model(scvi_model, unlabeled_category="Unknown")
scanvi_model.train()
SCANVI.from_scvi_model will also run setup_anndata. It will use the batch_key and layer used with SCVI, but here we add the labels_key.
For this part of the tutorial, we will create a new labels key in the reference anndata object to reflect the common scenario of having no labels for the query data.
SCANVI_LABELS_KEY = "labels_scanvi"
pancreas_ref.obs[SCANVI_LABELS_KEY] = pancreas_ref.obs["celltype"].values
Applying this workflow in the context of this tutorial:
# unlabeled category does not exist in adata.obs[labels_key]
# so all cells are treated as labeled
scanvi_ref = scvi.model.SCANVI.from_scvi_model(
scvi_ref,
unlabeled_category="Unknown",
labels_key=SCANVI_LABELS_KEY,
)
scanvi_ref.train(max_epochs=20, n_samples_per_label=100)
INFO Training for 20 epochs.
Epoch 1/20: 0%| | 0/20 [00:00<?, ?it/s]Epoch 2/20: 5%|▌ | 1/20 [00:01<00:20, 1.08s/it, v_num=1, train_loss_step=969, train_loss_epoch=896]Epoch 20/20: 100%|██████████| 20/20 [00:21<00:00, 1.11s/it, v_num=1, train_loss_step=811, train_loss_epoch=864] Epoch 20/20: 100%|██████████| 20/20 [00:21<00:00, 1.10s/it, v_num=1, train_loss_step=811, train_loss_epoch=864]
SCANVI_LATENT_KEY = "X_scANVI"
pancreas_ref.obsm[SCANVI_LATENT_KEY] = scanvi_ref.get_latent_representation()
sc.pp.neighbors(pancreas_ref, use_rep=SCANVI_LATENT_KEY)
sc.tl.leiden(pancreas_ref)
sc.tl.umap(pancreas_ref)
sc.pl.umap(
pancreas_ref,
color=["tech", "celltype"],
frameon=False,
ncols=1,
)

Update with query#
scanvi_ref_path = os.path.join(save_dir.name, "pancreas_scanvi_ref")
scanvi_ref.save(scanvi_ref_path, overwrite=True)
# again a no-op in this tutorial, but good practice to use
scvi.model.SCANVI.prepare_query_anndata(pancreas_query, scanvi_ref_path)
INFO File /tmp/tmp2qkhcse5/pancreas_scanvi_ref/model.pt already downloaded
INFO Found 100.0% reference vars in query data.
Notice that adata_query.obs["labels_scanvi"] does not exist. The load_query_data method detects this and fills it in adata_query with the unlabeled category (here "Unknown").
scanvi_query = scvi.model.SCANVI.load_query_data(pancreas_query, scanvi_ref_path)
INFO File /tmp/tmp2qkhcse5/pancreas_scanvi_ref/model.pt already downloaded
scanvi_query.train(
max_epochs=100,
plan_kwargs={"weight_decay": 0.0},
check_val_every_n_epoch=10,
)
INFO Training for 100 epochs.
Epoch 1/100: 0%| | 0/100 [00:00<?, ?it/s]Epoch 2/100: 1%| | 1/100 [00:00<00:30, 3.21it/s, v_num=1, train_loss_step=2e+3, train_loss_epoch=1.95e+3]Epoch 11/100: 10%|█ | 10/100 [00:03<00:29, 3.05it/s, v_num=1, train_loss_step=1.77e+3, train_loss_epoch=1.81e+3]Epoch 100/100: 100%|██████████| 100/100 [00:31<00:00, 3.02it/s, v_num=1, train_loss_step=1.76e+3, train_loss_epoch=1.75e+3]Epoch 100/100: 100%|██████████| 100/100 [00:31<00:00, 3.19it/s, v_num=1, train_loss_step=1.76e+3, train_loss_epoch=1.75e+3]
SCANVI_PREDICTIONS_KEY = "predictions_scanvi"
pancreas_query.obsm[SCANVI_LATENT_KEY] = scanvi_query.get_latent_representation()
pancreas_query.obs[SCANVI_PREDICTIONS_KEY] = scanvi_query.predict()
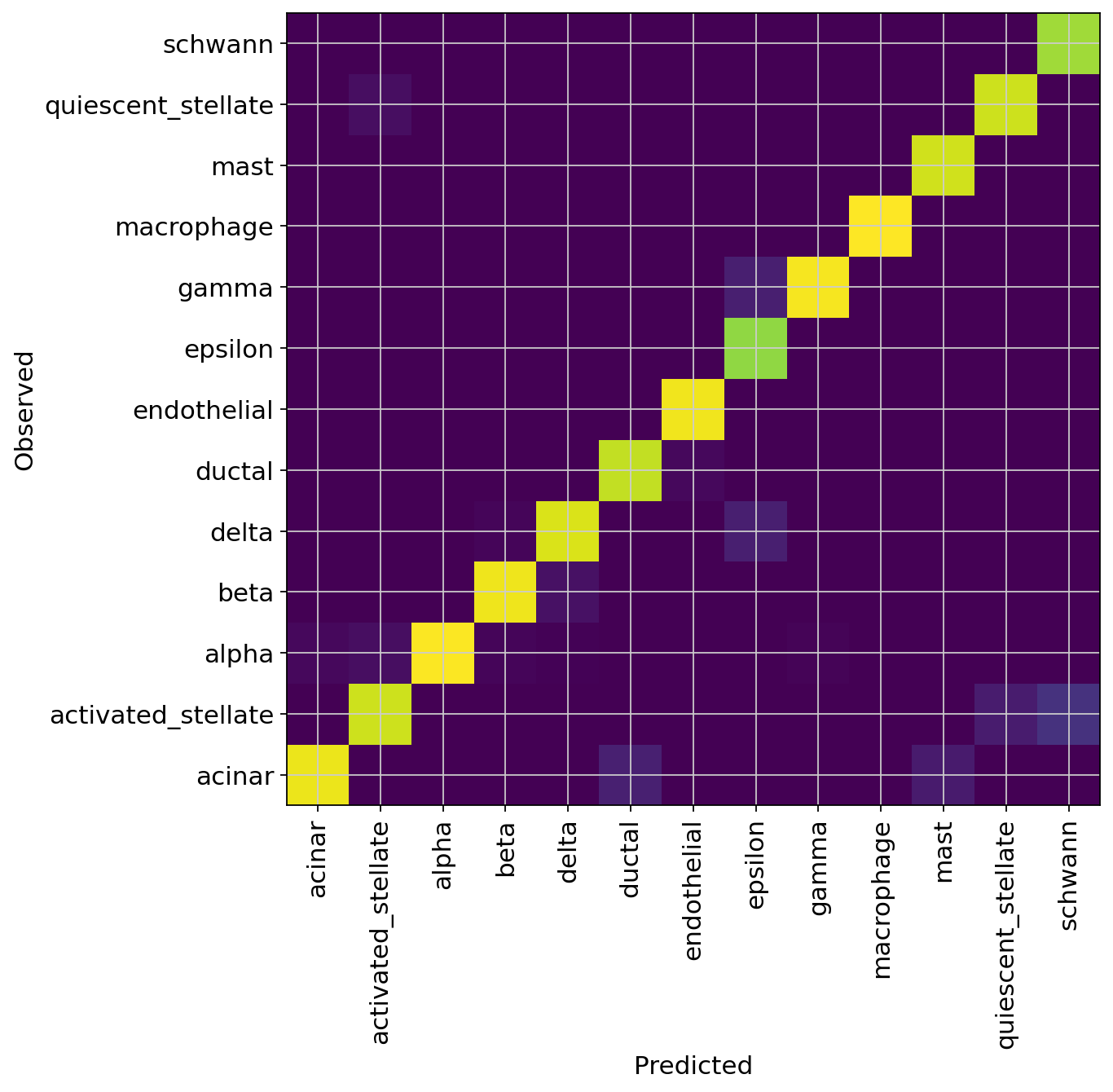
df = (
pancreas_query.obs.groupby(["celltype", SCANVI_PREDICTIONS_KEY])
.size()
.unstack(fill_value=0)
)
norm_df = df / df.sum(axis=0)
plt.figure(figsize=(8, 8))
_ = plt.pcolor(norm_df)
_ = plt.xticks(np.arange(0.5, len(df.columns), 1), df.columns, rotation=90)
_ = plt.yticks(np.arange(0.5, len(df.index), 1), df.index)
plt.xlabel("Predicted")
plt.ylabel("Observed")
Text(0, 0.5, 'Observed')
Analyze reference and query#
pancreas_full = anndata.concat([pancreas_query, pancreas_ref], label="batch")
pancreas_full
AnnData object with n_obs × n_vars = 16382 × 2000
obs: 'tech', 'celltype', 'size_factors', '_scvi_batch', '_scvi_labels', 'leiden', 'labels_scanvi', 'batch'
obsm: 'X_scVI', 'X_umap', 'X_scANVI'
layers: 'counts'
This just makes a column in the anndata corresponding to if the data come from the reference or query sets.
pancreas_full.obs["batch"] = pancreas_full.obs["batch"].cat.rename_categories(
["Query", "Reference"]
)
full_predictions = scanvi_query.predict(pancreas_full)
print(f"Acc: {np.mean(full_predictions == pancreas_full.obs['celltype'])}")
pancreas_full.obs[SCANVI_PREDICTIONS_KEY] = full_predictions
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
Acc: 0.9717372726162862
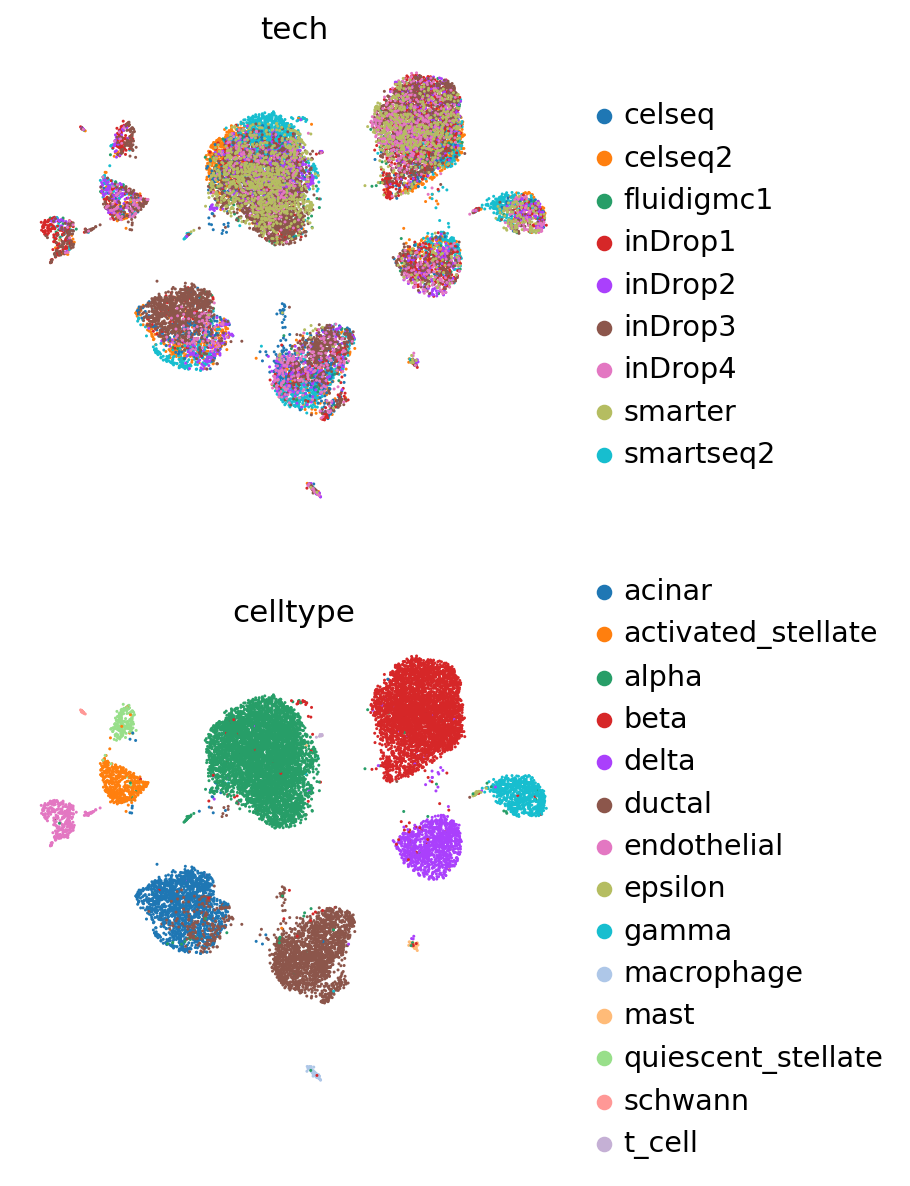
sc.pp.neighbors(pancreas_full, use_rep=SCANVI_LATENT_KEY)
sc.tl.leiden(pancreas_full)
sc.tl.umap(pancreas_full)
sc.pl.umap(
pancreas_full,
color=["tech", "celltype"],
frameon=False,
ncols=1,
)
ax = sc.pl.umap(
pancreas_full,
frameon=False,
show=False,
)
sc.pl.umap(
pancreas_full[: pancreas_query.n_obs],
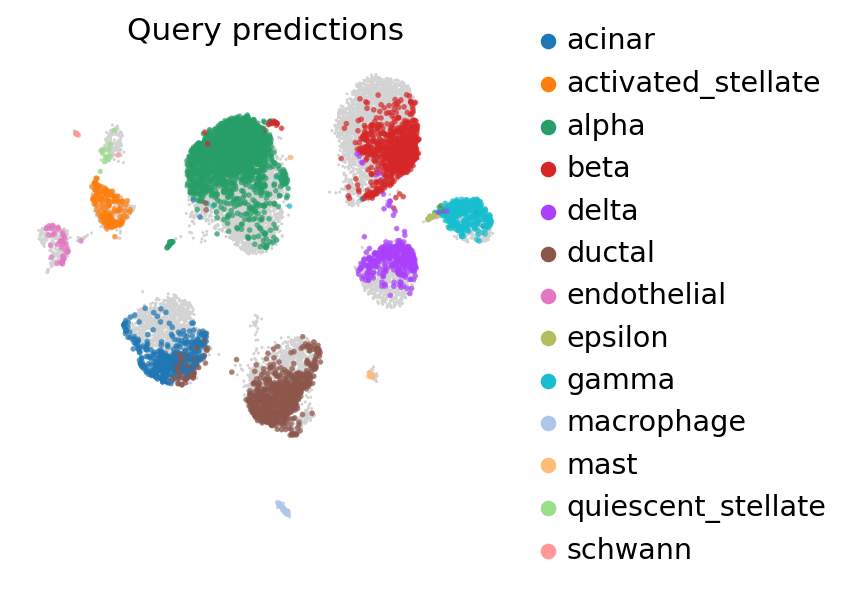
color=[SCANVI_PREDICTIONS_KEY],
frameon=False,
title="Query predictions",
ax=ax,
alpha=0.7,
)
ax = sc.pl.umap(
pancreas_full,
frameon=False,
show=False,
)
sc.pl.umap(
pancreas_full[: pancreas_query.n_obs],
color=["celltype"],
frameon=False,
title="Query observed cell types",
ax=ax,
alpha=0.7,
)

Reference mapping with TOTALVI#
This workflow works very similarly for TOTALVI. Here we demonstrate how to build a CITE-seq reference and use scRNA-seq only data as the query.
Assemble data#
For totalVI, we will treat two CITE-seq PBMC datasets from 10X Genomics as the reference. These datasets were already filtered for outliers like doublets, as described in the totalVI manuscript. There are 14 proteins in the reference.
pbmc_ref = scvi.data.pbmcs_10x_cite_seq(save_path=save_dir.name)
INFO Downloading file at /tmp/tmp2qkhcse5/pbmc_10k_protein_v3.h5ad
Downloading...: 24938it [00:00, 99295.94it/s]
INFO Downloading file at /tmp/tmp2qkhcse5/pbmc_5k_protein_v3.h5ad
Downloading...: 100%|██████████| 18295/18295.0 [00:00<00:00, 89430.22it/s]
In general, there will be some necessary data wrangling. For example, we need to provide totalVI with some protein data – and when it’s all zeros, totalVI identifies that the protein data is missing in this “batch”.
It could have also been the case that only some of the protein data was missing, in which case we would add zeros for each of the missing proteins.
pbmc_query = scvi.data.dataset_10x("pbmc_10k_v3", save_path=save_dir.name)
pbmc_query.obs["batch"] = "PBMC 10k (RNA only)"
# put matrix of zeros for protein expression (considered missing)
pro_exp = pbmc_ref.obsm["protein_expression"]
data = np.zeros((pbmc_query.n_obs, pro_exp.shape[1]))
pbmc_query.obsm["protein_expression"] = pd.DataFrame(
columns=pro_exp.columns, index=pbmc_query.obs_names, data=data
)
INFO Downloading file at /tmp/tmp2qkhcse5/pbmc_10k_v3/filtered_feature_bc_matrix.h5
Downloading...: 37492it [00:02, 15069.46it/s]
We do some light QC filtering on the query dataset (doublets, mitochondrial, etc.)
scrub = scr.Scrublet(pbmc_query.X)
doublet_scores, predicted_doublets = scrub.scrub_doublets()
pbmc_query = pbmc_query[~predicted_doublets].copy()
pbmc_query.var["mt"] = pbmc_query.var_names.str.startswith(
"MT-"
) # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(
pbmc_query, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
pbmc_query = pbmc_query[pbmc_query.obs.pct_counts_mt < 15, :].copy()
Preprocessing...
Simulating doublets...
Embedding transcriptomes using PCA...
Calculating doublet scores...
Automatically set threshold at doublet score = 0.32
Detected doublet rate = 4.9%
Estimated detectable doublet fraction = 56.4%
Overall doublet rate:
Expected = 10.0%
Estimated = 8.8%
Elapsed time: 10.2 seconds
Now to concatenate the objects, which intersects the genes properly.
pbmc_full = anndata.concat([pbmc_ref, pbmc_query])
And split them back up into reference and query (but now genes are properly aligned between objects).
pbmc_ref = pbmc_full[
np.logical_or(pbmc_full.obs.batch == "PBMC5k", pbmc_full.obs.batch == "PBMC10k")
].copy()
pbmc_query = pbmc_full[pbmc_full.obs.batch == "PBMC 10k (RNA only)"].copy()
We run gene selection on the reference, because that’s all that will be avaialble to us at first.
sc.pp.highly_variable_genes(
pbmc_ref,
n_top_genes=4000,
flavor="seurat_v3",
batch_key="batch",
subset=True,
)
Finally, we use these selected genes for the query dataset as well.
pbmc_query = pbmc_query[:, pbmc_ref.var_names].copy()
Train reference#
scvi.model.TOTALVI.setup_anndata(
pbmc_ref, batch_key="batch", protein_expression_obsm_key="protein_expression"
)
INFO Using column names from columns of adata.obsm['protein_expression']
totalvi_ref = scvi.model.TOTALVI(pbmc_ref, use_layer_norm="both", use_batch_norm="none")
INFO Computing empirical prior initialization for protein background.
totalvi_ref.train()
Epoch 16/400: 4%|▍ | 15/400 [00:06<02:32, 2.52it/s, v_num=1, train_loss_step=1.2e+3, train_loss_epoch=1.27e+3] Epoch 400/400: 100%|██████████| 400/400 [02:42<00:00, 2.42it/s, v_num=1, train_loss_step=1.23e+3, train_loss_epoch=1.23e+3]Epoch 400/400: 100%|██████████| 400/400 [02:42<00:00, 2.47it/s, v_num=1, train_loss_step=1.23e+3, train_loss_epoch=1.23e+3]
TOTALVI_LATENT_KEY = "X_totalVI"
pbmc_ref.obsm[TOTALVI_LATENT_KEY] = totalvi_ref.get_latent_representation()
sc.pp.neighbors(pbmc_ref, use_rep=TOTALVI_LATENT_KEY)
sc.tl.umap(pbmc_ref, min_dist=0.4)
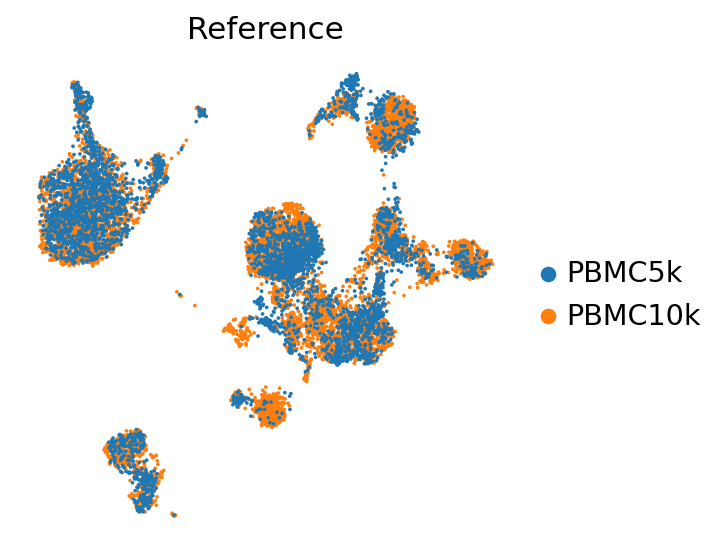
sc.pl.umap(pbmc_ref, color=["batch"], frameon=False, ncols=1, title="Reference")
totalvi_ref_path = os.path.join(save_dir.name, "pbmc_totalvi_ref")
totalvi_ref.save(totalvi_ref_path, overwrite=True)
Update with query#
scvi.model.TOTALVI.prepare_query_anndata(pbmc_query, totalvi_ref_path)
totalvi_query = scvi.model.TOTALVI.load_query_data(
pbmc_query,
totalvi_ref_path,
)
INFO File /tmp/tmp2qkhcse5/pbmc_totalvi_ref/model.pt already downloaded
INFO Found 100.0% reference vars in query data.
INFO File /tmp/tmp2qkhcse5/pbmc_totalvi_ref/model.pt already downloaded
INFO Found batches with missing protein expression
INFO Computing empirical prior initialization for protein background.
totalvi_query.train(200, plan_kwargs={"weight_decay": 0.0})
Epoch 7/200: 3%|▎ | 6/200 [00:02<01:33, 2.08it/s, v_num=1, train_loss_step=1.53e+3, train_loss_epoch=1.6e+3] Epoch 147/200: 73%|███████▎ | 146/200 [01:07<00:23, 2.28it/s, v_num=1, train_loss_step=1.58e+3, train_loss_epoch=1.51e+3]Epoch 00147: reducing learning rate of group 0 to 2.4000e-03.
Epoch 161/200: 80%|████████ | 161/200 [01:14<00:18, 2.17it/s, v_num=1, train_loss_step=1.49e+3, train_loss_epoch=1514.25]
Monitored metric elbo_validation did not improve in the last 45 records. Best score: 1500.559. Signaling Trainer to stop.
pbmc_query.obsm[TOTALVI_LATENT_KEY] = totalvi_query.get_latent_representation()
sc.pp.neighbors(pbmc_query, use_rep=TOTALVI_LATENT_KEY)
sc.tl.umap(pbmc_query, min_dist=0.4)
Impute protein data for query and visualize#
Now that we have updated with the query, we can impute the proteins that were observed in the reference, using the transform_batch parameter.
_, imputed_proteins = totalvi_query.get_normalized_expression(
pbmc_query,
n_samples=10,
return_mean=True,
transform_batch=["PBMC10k", "PBMC5k"],
)
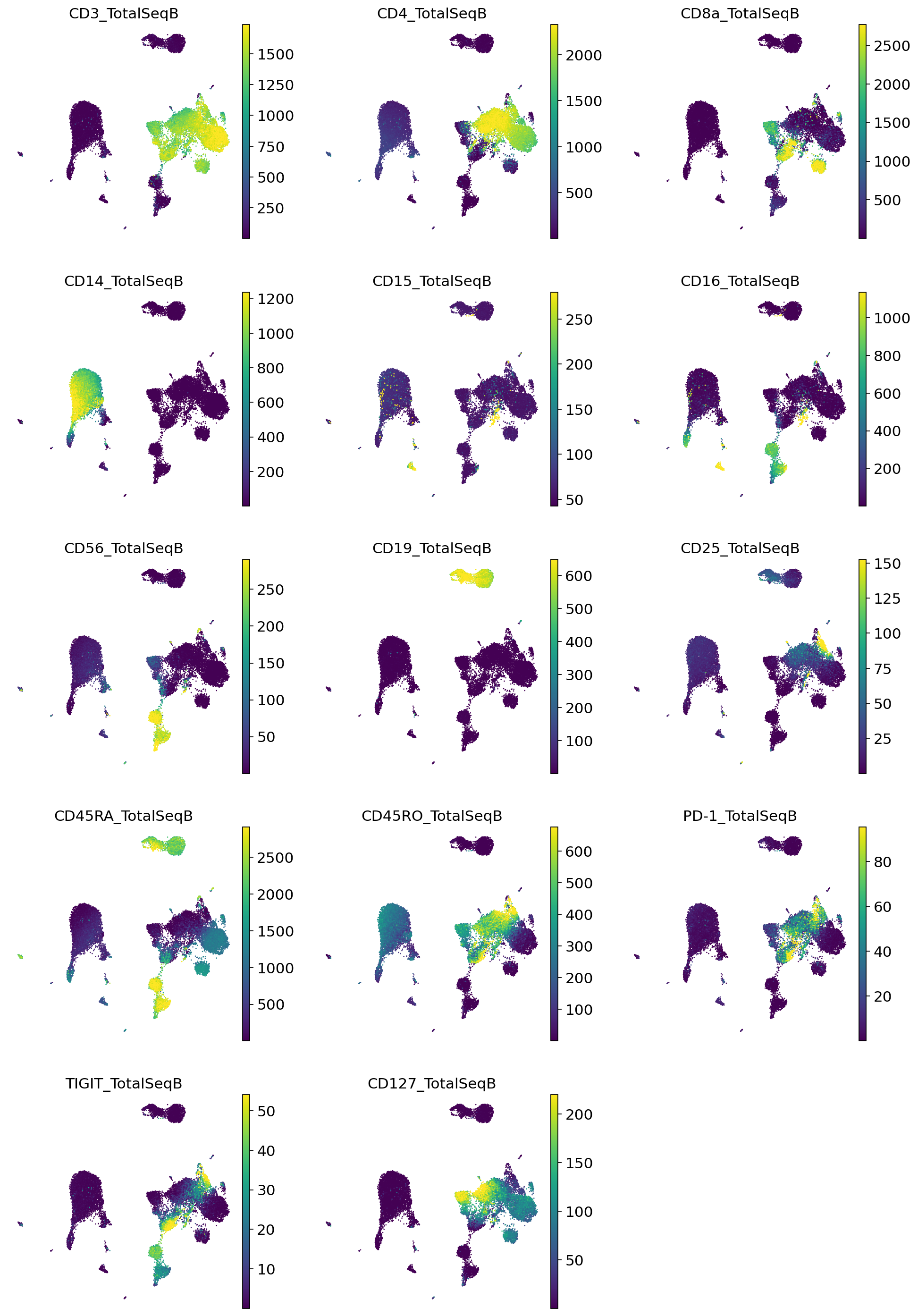
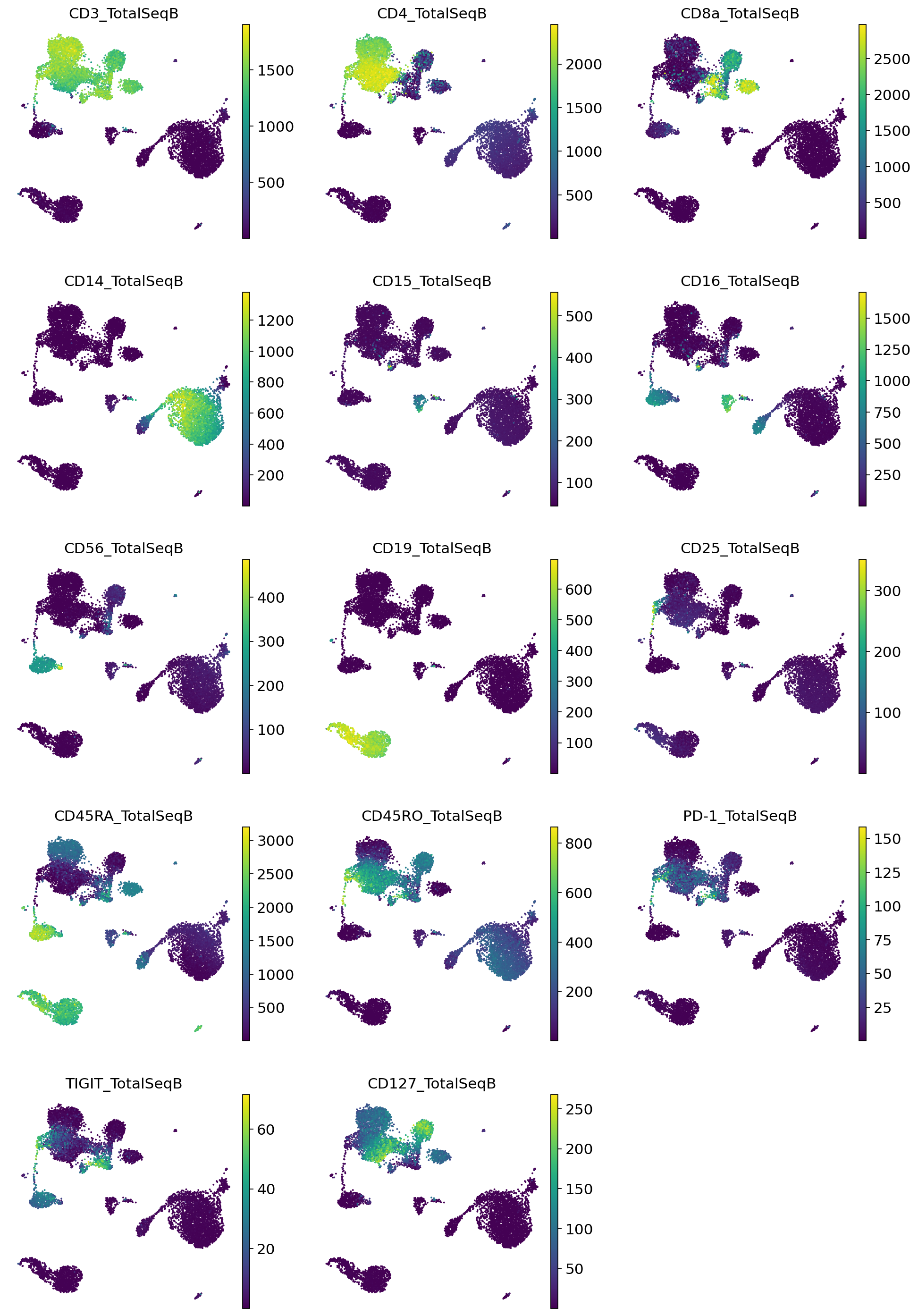
Very quickly we can identify the major expected subpopulations of B cells, CD4 T cells, CD8 T cells, monocytes, etc.
pbmc_query.obs = pd.concat([pbmc_query.obs, imputed_proteins], axis=1)
sc.pl.umap(
pbmc_query,
color=imputed_proteins.columns,
frameon=False,
ncols=3,
)
Visualize reference and query#
pbmc_full = anndata.concat([pbmc_query, pbmc_ref])
pbmc_full.obsm[TOTALVI_LATENT_KEY] = totalvi_query.get_latent_representation(pbmc_full)
sc.pp.neighbors(pbmc_full, use_rep=TOTALVI_LATENT_KEY)
sc.tl.umap(pbmc_full, min_dist=0.3)
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
INFO Found batches with missing protein expression
_, imputed_proteins_all = totalvi_query.get_normalized_expression(
pbmc_full,
n_samples=10,
return_mean=True,
transform_batch=["PBMC10k", "PBMC5k"],
)
for i, p in enumerate(imputed_proteins_all.columns):
pbmc_full.obs[p] = imputed_proteins_all[p].to_numpy().copy()
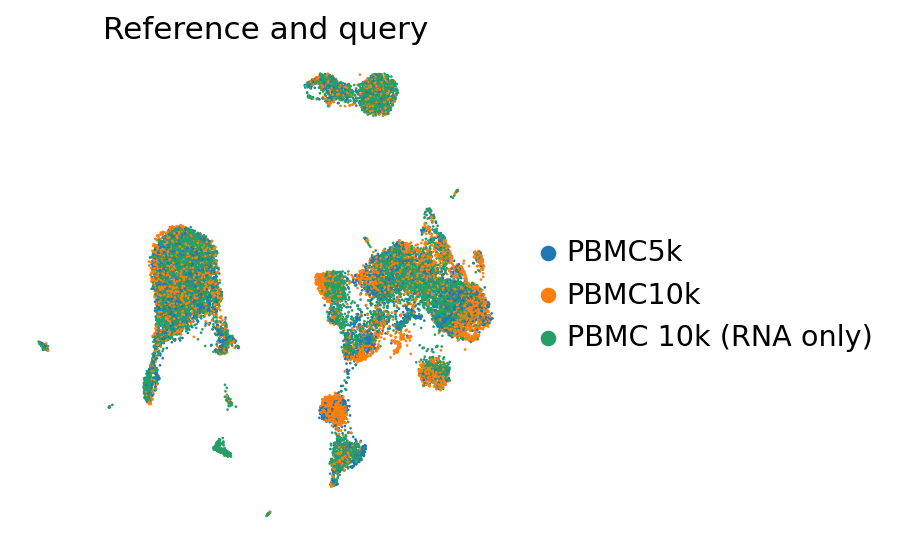
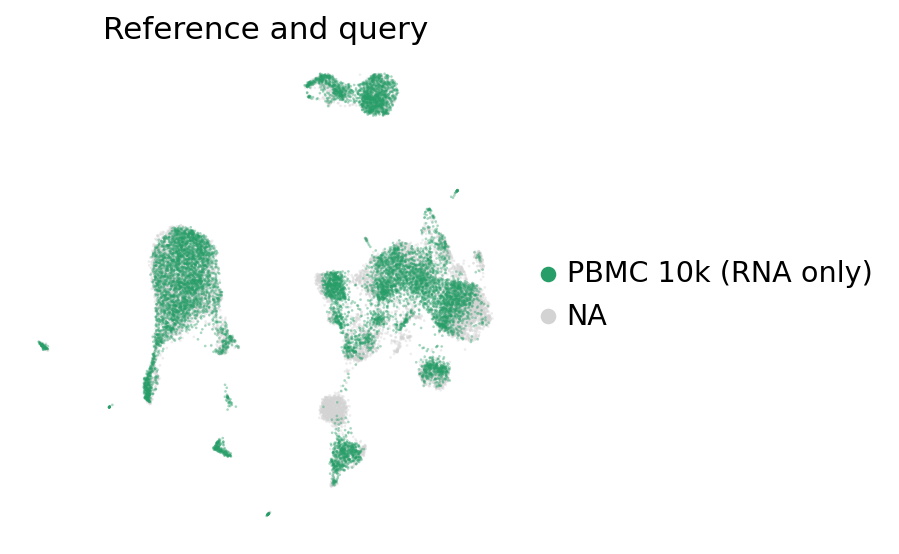
perm_inds = np.random.permutation(np.arange(pbmc_full.n_obs))
sc.pl.umap(
pbmc_full[perm_inds],
color=["batch"],
frameon=False,
ncols=1,
title="Reference and query",
)
ax = sc.pl.umap(
pbmc_full,
color="batch",
groups=["PBMC 10k (RNA only)"],
frameon=False,
ncols=1,
title="Reference and query",
alpha=0.4,
)
sc.pl.umap(
pbmc_full,
color=imputed_proteins_all.columns,
frameon=False,
ncols=3,
vmax="p99",
)