Querying the Human Lung Cell Atlas#
Here we demonstrate how to query the Human Lung Cell Atlas using scANVI, scArches, and scvi-hub.
Sikkema, Lisa, et al. “An integrated cell atlas of the human lung in health and disease.” bioRxiv (2022).
If you use this tutorial in your research we recommend citing the HLCA as well as scANVI, scArches, and scvi-tools, which can be found on the references page at Gayoso22, Lotfollahi21, Xu21 respectively.
This tutorial is adapted from a similar one presented by the HLCA authors.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
WARNING: Running pip as the 'root' user can result in broken permissions and conflicting behaviour with the system package manager. It is recommended to use a virtual environment instead: https://pip.pypa.io/warnings/venv
import os
import tempfile
import anndata
import numba
import numpy as np
import pandas as pd
import pooch
import pynndescent
import scanpy as sc
import scvi
import seaborn as sns
import torch
from scvi.hub import HubModel
from scvi.model.utils import mde
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.1.0
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Download the reference files#
First we download the pre-trained scANVI model from the HuggingFace repo.
hubmodel = HubModel.pull_from_huggingface_hub(
"scvi-tools/human-lung-cell-atlas", cache_dir=save_dir.name
)
adata = hubmodel.adata
model = hubmodel.model
INFO Reading adata...
INFO Loading model...
INFO File
/tmp/tmpmw6gbji7/models--scvi-tools--human-lung-cell-atlas/snapshots/7af8f610b2c46b715e62cf46bc14163d8a3e6
79e/model.pt already downloaded
adata
AnnData object with n_obs × n_vars = 584944 × 2000
obs: 'suspension_type', 'donor_id', 'is_primary_data', 'assay_ontology_term_id', 'cell_type_ontology_term_id', 'development_stage_ontology_term_id', 'disease_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'tissue_ontology_term_id', 'organism_ontology_term_id', 'sex_ontology_term_id', 'BMI', 'age_or_mean_of_age_range', 'age_range', 'anatomical_region_ccf_score', 'ann_coarse_for_GWAS_and_modeling', 'ann_finest_level', 'ann_level_1', 'ann_level_2', 'ann_level_3', 'ann_level_4', 'ann_level_5', 'cause_of_death', 'dataset', 'entropy_dataset_leiden_3', 'entropy_original_ann_level_1_leiden_3', 'entropy_original_ann_level_2_clean_leiden_3', 'entropy_original_ann_level_3_clean_leiden_3', 'entropy_subject_ID_leiden_3', 'fresh_or_frozen', 'leiden_1', 'leiden_2', 'leiden_3', 'leiden_4', 'leiden_5', 'log10_total_counts', 'lung_condition', 'mixed_ancestry', 'n_genes_detected', 'original_ann_highest_res', 'original_ann_level_1', 'original_ann_level_2', 'original_ann_level_3', 'original_ann_level_4', 'original_ann_level_5', 'original_ann_nonharmonized', 'reannotation_type', 'reference_genome', 'sample', 'scanvi_label', 'sequencing_platform', 'size_factors', 'smoking_status', 'study', 'subject_type', 'tissue_dissociation_protocol', 'tissue_level_2', 'tissue_level_3', 'tissue_sampling_method', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', '_scvi_batch', '_scvi_labels', '_scanvi_observed_lib_size'
var: 'feature_name'
uns: '_scvi_adata_minify_type', '_scvi_manager_uuid', '_scvi_uuid'
obsm: 'SCANVI_latent_qzm', 'SCANVI_latent_qzv', 'X_scanvi_emb', 'X_umap', '_scanvi_latent_qzm', '_scanvi_latent_qzv'
obsp: 'connectivities', 'distances'
model
ScanVI Model with the following params: unlabeled_category: unlabeled, n_hidden: 128, n_latent: 30, n_layers: 2, dropout_rate: 0.1, dispersion: gene, gene_likelihood: nb Training status: Trained Model's adata is minified?: True
model.view_anndata_setup()
Anndata setup with scvi-tools version 1.1.0.
Setup via `SCANVI.setup_anndata` with arguments:
{ │ 'labels_key': 'scanvi_label', │ 'unlabeled_category': 'unlabeled', │ 'layer': None, │ 'batch_key': 'dataset', │ 'size_factor_key': None, │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None }
Summary Statistics ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━┓ ┃ Summary Stat Key ┃ Value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━┩ │ n_batch │ 14 │ │ n_cells │ 584944 │ │ n_extra_categorical_covs │ 0 │ │ n_extra_continuous_covs │ 0 │ │ n_labels │ 29 │ │ n_latent_qzm │ 30 │ │ n_latent_qzv │ 30 │ │ n_vars │ 2000 │ └──────────────────────────┴────────┘
Data Registry ┏━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Registry Key ┃ scvi-tools Location ┃ ┡━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ X │ adata.X │ │ batch │ adata.obs['_scvi_batch'] │ │ labels │ adata.obs['_scvi_labels'] │ │ latent_qzm │ adata.obsm['_scanvi_latent_qzm'] │ │ latent_qzv │ adata.obsm['_scanvi_latent_qzv'] │ │ minify_type │ adata.uns['_scvi_adata_minify_type'] │ │ observed_lib_size │ adata.obs['_scanvi_observed_lib_size'] │ └───────────────────┴────────────────────────────────────────┘
batch State Registry ┏━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['dataset'] │ Banovich_Kropski_2020 │ 0 │ │ │ Barbry_Leroy_2020 │ 1 │ │ │ Jain_Misharin_2021_10Xv1 │ 2 │ │ │ Jain_Misharin_2021_10Xv2 │ 3 │ │ │ Krasnow_2020 │ 4 │ │ │ Lafyatis_Rojas_2019_10Xv1 │ 5 │ │ │ Lafyatis_Rojas_2019_10Xv2 │ 6 │ │ │ Meyer_2019 │ 7 │ │ │ Misharin_2021 │ 8 │ │ │ Misharin_Budinger_2018 │ 9 │ │ │ Nawijn_2021 │ 10 │ │ │ Seibold_2020_10Xv2 │ 11 │ │ │ Seibold_2020_10Xv3 │ 12 │ │ │ Teichmann_Meyer_2019 │ 13 │ └──────────────────────┴───────────────────────────┴─────────────────────┘
labels State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['scanvi_label'] │ AT1 │ 0 │ │ │ AT2 │ 1 │ │ │ Arterial EC │ 2 │ │ │ B cell lineage │ 3 │ │ │ Basal │ 4 │ │ │ Bronchial Vessel 1 │ 5 │ │ │ Bronchial Vessel 2 │ 6 │ │ │ Capillary │ 7 │ │ │ Ciliated │ 8 │ │ │ Dendritic cells │ 9 │ │ │ Fibroblast lineage │ 10 │ │ │ KRT5- KRT17+ epithelial │ 11 │ │ │ Lymphatic EC │ 12 │ │ │ Macrophages │ 13 │ │ │ Mast cells │ 14 │ │ │ Megakaryocytes │ 15 │ │ │ Mesothelium │ 16 │ │ │ Monocytes │ 17 │ │ │ Neutrophilic │ 18 │ │ │ Non-T/B cells │ 19 │ │ │ Proliferating cells │ 20 │ │ │ Rare │ 21 │ │ │ Secretory │ 22 │ │ │ Smooth Muscle │ 23 │ │ │ Squamous │ 24 │ │ │ Submucosal Secretory │ 25 │ │ │ T cell lineage │ 26 │ │ │ Venous │ 27 │ │ │ unlabeled │ 28 │ └───────────────────────────┴─────────────────────────┴─────────────────────┘
Learn a neighbors index on reference latent space#
Here we create the neighbors index using PyNNDescent. We will use this later to classify query cells. PyNNDescent is an extremely fast approximate neighbors technique.
In the case of the HubModel instance above, we see that the data is in minified mode, meaning the count data is not actually in the object, and we only store a minified representation of the data. We see that we can access the mean of the embedding (latent_qzm) above.
X_train = adata.obsm["_scanvi_latent_qzm"]
ref_nn_index = pynndescent.NNDescent(X_train)
ref_nn_index.prepare()
Download query data#
In this tutorial we use the fresh, single-cell sample from the following publication:
Delorey, Toni M., et al. “COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets.” Nature 595.7865 (2021): 107-113.
In principle at this stage you may load your own data. There are few important notes though:
Using the HLCA requires using Gene IDs for the query data
The query data should include batches in
query_data.obs["dataset"]It’s necessary to run
query_data.obs["scanvi_label"] = "unlabeled"so that scvi-tools can properly register the query data.
def download_data(save_path: str):
"""Download and cache the query data."""
data_path = pooch.retrieve(
url="https://ftp.ncbi.nlm.nih.gov/geo/samples/GSM5230nnn/GSM5230027/suppl/GSM5230027_04-P103142-S149-R01_raw_feature_bc_matrix.h5.gz",
known_hash="3b7f8318059d655ea774752b6d8b13381323f5018e9f3868ffc53674f94f537f",
fname="query_data.h5.gz",
path=save_path,
processor=pooch.Decompress(),
progressbar=True,
)
metadata_path = pooch.retrieve(
url="https://ftp.ncbi.nlm.nih.gov/geo/series/GSE171nnn/GSE171668/suppl/GSE171668_lung_metadata.csv.gz",
known_hash="290b0ac86e85183e65eefb68670ad27fc5156866144d9ac6f2eb27f34e31e79e",
fname="query_metadata.csv.gz",
path=save_path,
processor=pooch.Decompress(),
progressbar=True,
)
return data_path, metadata_path
query_data_path, query_metadata_path = download_data(save_dir.name)
query_adata = sc.read_10x_h5(query_data_path)
query_metadata = pd.read_csv(query_metadata_path, index_col=0)
# clean up .var.index (gene names)
query_adata.var["gene_names"] = query_adata.var.index
query_adata.var.index = [idx.split("___")[-1] for idx in query_adata.var.gene_ids]
# clean up cell barcodes:
query_adata.obs.index = query_adata.obs.index.str.rstrip("-1")
# read in metadata (to select only cells of interest and remove empty drops)
# subset to cells from our sample
query_metadata = query_metadata.loc[query_metadata.donor == "D12_4", :].copy()
# clean up barcodes:
query_metadata.index = [idx.split("-")[-1] for idx in query_metadata.index]
# subset adata to cells in metadata:
query_adata = query_adata[query_metadata.index, :].copy()
# add dataset information:
query_adata.obs["dataset"] = "test_dataset_delorey_regev"
Loading the query model from the reference files#
Here we run prepare_query_anndata, which reorders the genes and pads any missing genes with 0s. This should generally be run before reference mapping with scArches to ensure data correctness.
Important
Below we use the path to the model we downloaded from HuggingFace. While in most cases the model instance can be used instead of the path, here the reference model’s adata is in minified mode.
scvi.model.SCANVI.prepare_query_anndata(query_adata, model)
INFO Found 99.65% reference vars in query data.
From above, we see that the model is expecting a labels key with the name "scanvi_label".
query_adata.obs["scanvi_label"] = "unlabeled"
query_model = scvi.model.SCANVI.load_query_data(query_adata, model)
Here we use scArches/scANVI-specific query training arguments.
surgery_epochs = 500
train_kwargs_surgery = {
"early_stopping": True,
"early_stopping_monitor": "elbo_train",
"early_stopping_patience": 10,
"early_stopping_min_delta": 0.001,
"plan_kwargs": {"weight_decay": 0.0},
}
query_model.train(max_epochs=surgery_epochs, **train_kwargs_surgery)
INFO Training for 500 epochs.
Epoch 228/500: 46%|████▌ | 228/500 [00:30<00:36, 7.53it/s, v_num=1, train_loss_step=471, train_loss_epoch=482]
Monitored metric elbo_train did not improve in the last 10 records. Best score: 526.328. Signaling Trainer to stop.
query_model_path = os.path.join(save_dir.name, "query_model")
query_model.save(query_model_path, overwrite=True)
query_emb = anndata.AnnData(query_model.get_latent_representation())
query_emb.obs_names = query_adata.obs_names
Now let’s store the predictions in the query embedding object. We reuse the PyNNDescent index from before, converting distances to affinities, and weighting the predictions using these affinities.
This follows the same approach used in the HLCA.
ref_neighbors, ref_distances = ref_nn_index.query(query_emb.X)
# convert distances to affinities
stds = np.std(ref_distances, axis=1)
stds = (2.0 / stds) ** 2
stds = stds.reshape(-1, 1)
ref_distances_tilda = np.exp(-np.true_divide(ref_distances, stds))
weights = ref_distances_tilda / np.sum(ref_distances_tilda, axis=1, keepdims=True)
@numba.njit
def weighted_prediction(weights, ref_cats):
"""Get highest weight category."""
N = len(weights)
predictions = np.zeros((N,), dtype=ref_cats.dtype)
uncertainty = np.zeros((N,))
for i in range(N):
obs_weights = weights[i]
obs_cats = ref_cats[i]
best_prob = 0
for c in np.unique(obs_cats):
cand_prob = np.sum(obs_weights[obs_cats == c])
if cand_prob > best_prob:
best_prob = cand_prob
predictions[i] = c
uncertainty[i] = max(1 - best_prob, 0)
return predictions, uncertainty
# for each annotation level, get prediction and uncertainty
label_keys = [f"ann_level_{i}" for i in range(1, 6)] + ["ann_finest_level"]
for l in label_keys:
ref_cats = adata.obs[l].cat.codes.to_numpy()[ref_neighbors]
p, u = weighted_prediction(weights, ref_cats)
p = np.asarray(adata.obs[l].cat.categories)[p]
query_emb.obs[l + "_pred"], query_emb.obs[l + "_uncertainty"] = p, u
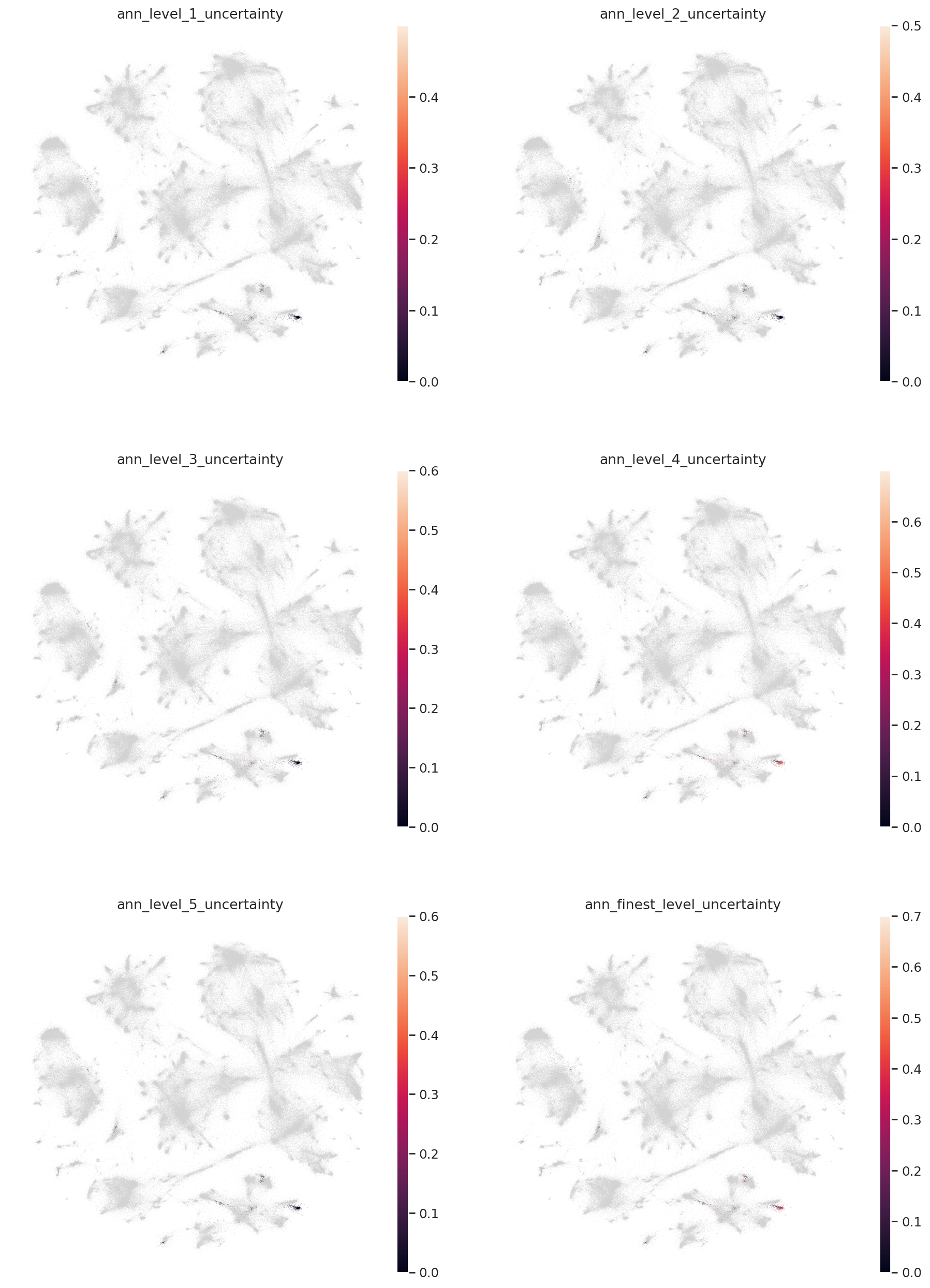
Now let’s filter our predictions on the uncertainty threshold, which is discussed in the HLCA manuscript.
uncertainty_threshold = 0.2
for l in label_keys:
mask = query_emb.obs[l + "_uncertainty"] > 0.2
print(f"{l}: {sum(mask)/len(mask)} unknown")
query_emb.obs[l + "_pred"].loc[mask] = "Unknown"
ann_level_1: 0.003919372900335946 unknown
ann_level_2: 0.007838745800671893 unknown
ann_level_3: 0.07334826427771557 unknown
ann_level_4: 0.38577827547592386 unknown
ann_level_5: 0.0083986562150056 unknown
ann_finest_level: 0.406494960806271 unknown
query_emb.obs["dataset"] = "test_dataset_delorey_regev"
Combine embeddings#
ref_emb = anndata.AnnData(X_train, obs=adata.obs)
ref_emb
AnnData object with n_obs × n_vars = 584944 × 30
obs: 'suspension_type', 'donor_id', 'is_primary_data', 'assay_ontology_term_id', 'cell_type_ontology_term_id', 'development_stage_ontology_term_id', 'disease_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'tissue_ontology_term_id', 'organism_ontology_term_id', 'sex_ontology_term_id', 'BMI', 'age_or_mean_of_age_range', 'age_range', 'anatomical_region_ccf_score', 'ann_coarse_for_GWAS_and_modeling', 'ann_finest_level', 'ann_level_1', 'ann_level_2', 'ann_level_3', 'ann_level_4', 'ann_level_5', 'cause_of_death', 'dataset', 'entropy_dataset_leiden_3', 'entropy_original_ann_level_1_leiden_3', 'entropy_original_ann_level_2_clean_leiden_3', 'entropy_original_ann_level_3_clean_leiden_3', 'entropy_subject_ID_leiden_3', 'fresh_or_frozen', 'leiden_1', 'leiden_2', 'leiden_3', 'leiden_4', 'leiden_5', 'log10_total_counts', 'lung_condition', 'mixed_ancestry', 'n_genes_detected', 'original_ann_highest_res', 'original_ann_level_1', 'original_ann_level_2', 'original_ann_level_3', 'original_ann_level_4', 'original_ann_level_5', 'original_ann_nonharmonized', 'reannotation_type', 'reference_genome', 'sample', 'scanvi_label', 'sequencing_platform', 'size_factors', 'smoking_status', 'study', 'subject_type', 'tissue_dissociation_protocol', 'tissue_level_2', 'tissue_level_3', 'tissue_sampling_method', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', '_scvi_batch', '_scvi_labels', '_scanvi_observed_lib_size'
query_emb
AnnData object with n_obs × n_vars = 1786 × 30
obs: 'ann_level_1_pred', 'ann_level_1_uncertainty', 'ann_level_2_pred', 'ann_level_2_uncertainty', 'ann_level_3_pred', 'ann_level_3_uncertainty', 'ann_level_4_pred', 'ann_level_4_uncertainty', 'ann_level_5_pred', 'ann_level_5_uncertainty', 'ann_finest_level_pred', 'ann_finest_level_uncertainty', 'dataset'
combined_emb = ref_emb.concatenate(query_emb)
Visualize embeddings and predictions#
To visualize here we use minimum distortion embeddings, which for now can be thought of as an alternative of UMAP that is GPU-accelerated (= really fast on Colab). While we use init="random" here, we recommend removing this argument in practice and only leave it here to make the notebook run faster.
MDE_KEY = "X_mde"
combined_emb.obsm[MDE_KEY] = mde(combined_emb.X, init="random")
INFO Using cuda:0 for `pymde.preserve_neighbors`.
combined_emb
AnnData object with n_obs × n_vars = 586730 × 30
obs: 'suspension_type', 'donor_id', 'is_primary_data', 'assay_ontology_term_id', 'cell_type_ontology_term_id', 'development_stage_ontology_term_id', 'disease_ontology_term_id', 'self_reported_ethnicity_ontology_term_id', 'tissue_ontology_term_id', 'organism_ontology_term_id', 'sex_ontology_term_id', 'BMI', 'age_or_mean_of_age_range', 'age_range', 'anatomical_region_ccf_score', 'ann_coarse_for_GWAS_and_modeling', 'ann_finest_level', 'ann_level_1', 'ann_level_2', 'ann_level_3', 'ann_level_4', 'ann_level_5', 'cause_of_death', 'dataset', 'entropy_dataset_leiden_3', 'entropy_original_ann_level_1_leiden_3', 'entropy_original_ann_level_2_clean_leiden_3', 'entropy_original_ann_level_3_clean_leiden_3', 'entropy_subject_ID_leiden_3', 'fresh_or_frozen', 'leiden_1', 'leiden_2', 'leiden_3', 'leiden_4', 'leiden_5', 'log10_total_counts', 'lung_condition', 'mixed_ancestry', 'n_genes_detected', 'original_ann_highest_res', 'original_ann_level_1', 'original_ann_level_2', 'original_ann_level_3', 'original_ann_level_4', 'original_ann_level_5', 'original_ann_nonharmonized', 'reannotation_type', 'reference_genome', 'sample', 'scanvi_label', 'sequencing_platform', 'size_factors', 'smoking_status', 'study', 'subject_type', 'tissue_dissociation_protocol', 'tissue_level_2', 'tissue_level_3', 'tissue_sampling_method', 'cell_type', 'assay', 'disease', 'organism', 'sex', 'tissue', 'self_reported_ethnicity', 'development_stage', '_scvi_batch', '_scvi_labels', '_scanvi_observed_lib_size', 'ann_level_1_pred', 'ann_level_1_uncertainty', 'ann_level_2_pred', 'ann_level_2_uncertainty', 'ann_level_3_pred', 'ann_level_3_uncertainty', 'ann_level_4_pred', 'ann_level_4_uncertainty', 'ann_level_5_pred', 'ann_level_5_uncertainty', 'ann_finest_level_pred', 'ann_finest_level_uncertainty', 'batch'
obsm: 'X_mde'
colors = [l + "_uncertainty" for l in label_keys]
sc.pl.embedding(
combined_emb,
basis=MDE_KEY,
color=colors,
ncols=2,
)
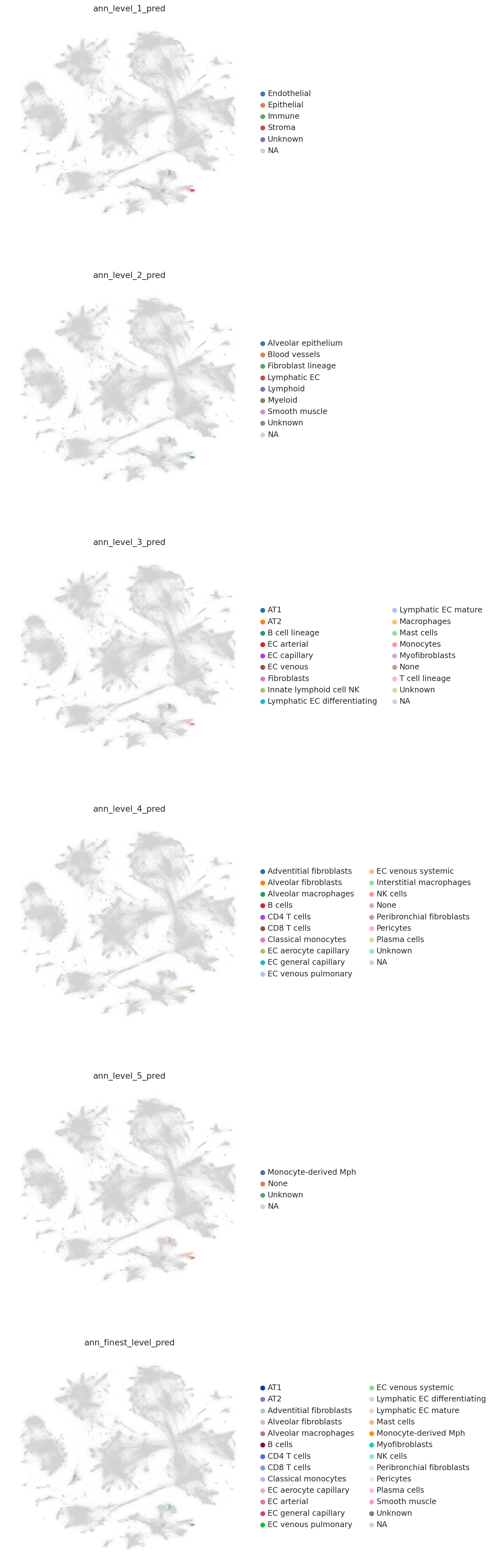
colors = [l + "_pred" for l in label_keys]
sc.pl.embedding(combined_emb, basis=MDE_KEY, color=colors, ncols=1, size=0.5)
sc.pl.embedding(
combined_emb, basis=MDE_KEY, color="ann_finest_level", ncols=1, size=0.5
)
