Spatial mapping with Tangram#
This tutorial demonstrates how to use Tangram (original code) to map spatial transcriptomics data. Here, we follow the corresponding tutorial at Squidpy. We do not provide much explanation, and instead refer to the original tutorial.
In scvi-tools, the "cells" and "constrained" modes of Tangram are implemented. In the "cells" mode, the priors will need to be calculated manually. We show this below.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import tempfile
import matplotlib.pyplot as plt
import mudata
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import seaborn as sns
import squidpy as sq
from scvi.external import Tangram
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.2
Note
You can modify save_dir below to change where the data files for this tutorial are saved.
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Load and preprocess data with squidpy#
adata_sp = sq.datasets.visium_fluo_adata_crop("data/visium_fluo_adata_crop.h5ad")
adata_sp = adata_sp[adata_sp.obs.cluster.isin([f"Cortex_{i}" for i in np.arange(1, 5)])].copy()
img = sq.datasets.visium_fluo_image_crop("data")
INFO Downloading data.tiff from https://exampledata.scverse.org/squidpy/figshare/visium_fluo_image_crop.tiff
adata_sc = sq.datasets.sc_mouse_cortex("data/sc_mouse_cortex.h5ad")
INFO Downloading sc_mouse_cortex.h5ad from
https://exampledata.scverse.org/squidpy/figshare/sc_mouse_cortex.h5ad
sc.pp.filter_genes(adata_sp, min_cells=1)
sc.pp.filter_genes(adata_sc, min_cells=1)
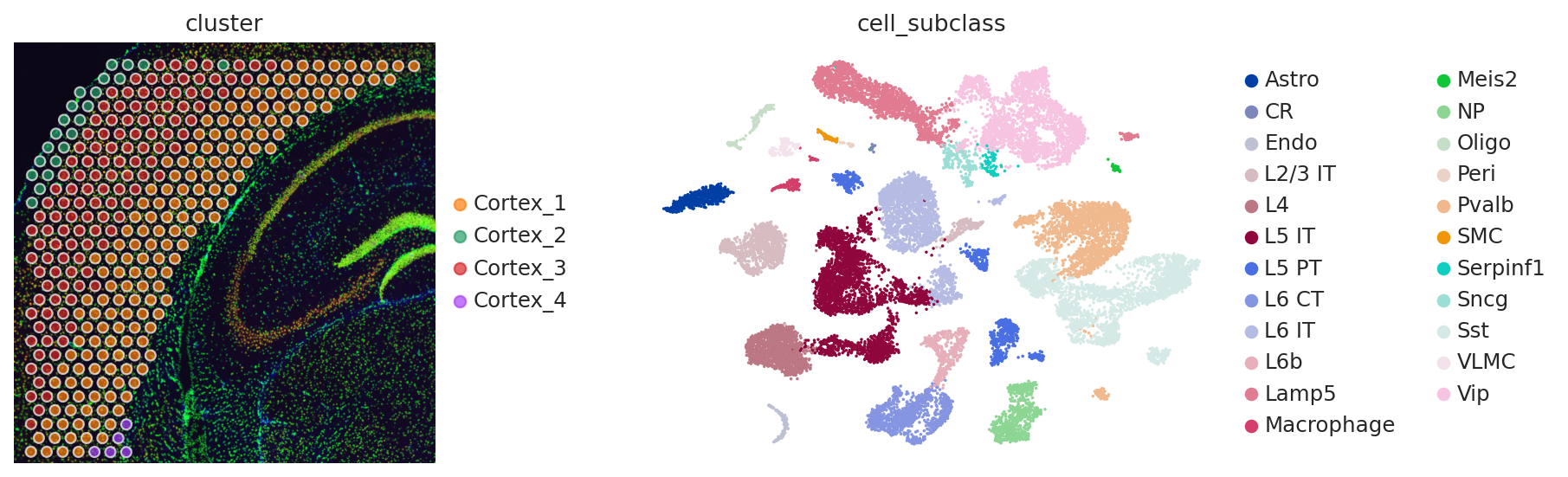
fig, axs = plt.subplots(1, 2, figsize=(12, 4))
sq.pl.spatial_scatter(adata_sp, color="cluster", alpha=0.7, frameon=False, ax=axs[0])
sc.pl.umap(adata_sc, color="cell_subclass", size=10, frameon=False, ax=axs[1])
mdata = mudata.MuData(
{
"sp": adata_sp,
"sc": adata_sc,
}
)
sq.im.process(img=img, layer="image", method="smooth")
sq.im.segment(
img=img,
layer="image_smooth",
method="watershed",
channel=0,
)
# define image layer to use for segmentation
features_kwargs = {
"segmentation": {
"label_layer": "segmented_watershed",
"props": ["label", "centroid"],
"channels": [1, 2],
}
}
# calculate segmentation features
sq.im.calculate_image_features(
adata_sp,
img,
layer="image",
key_added="image_features",
features_kwargs=features_kwargs,
features="segmentation",
mask_circle=True,
)
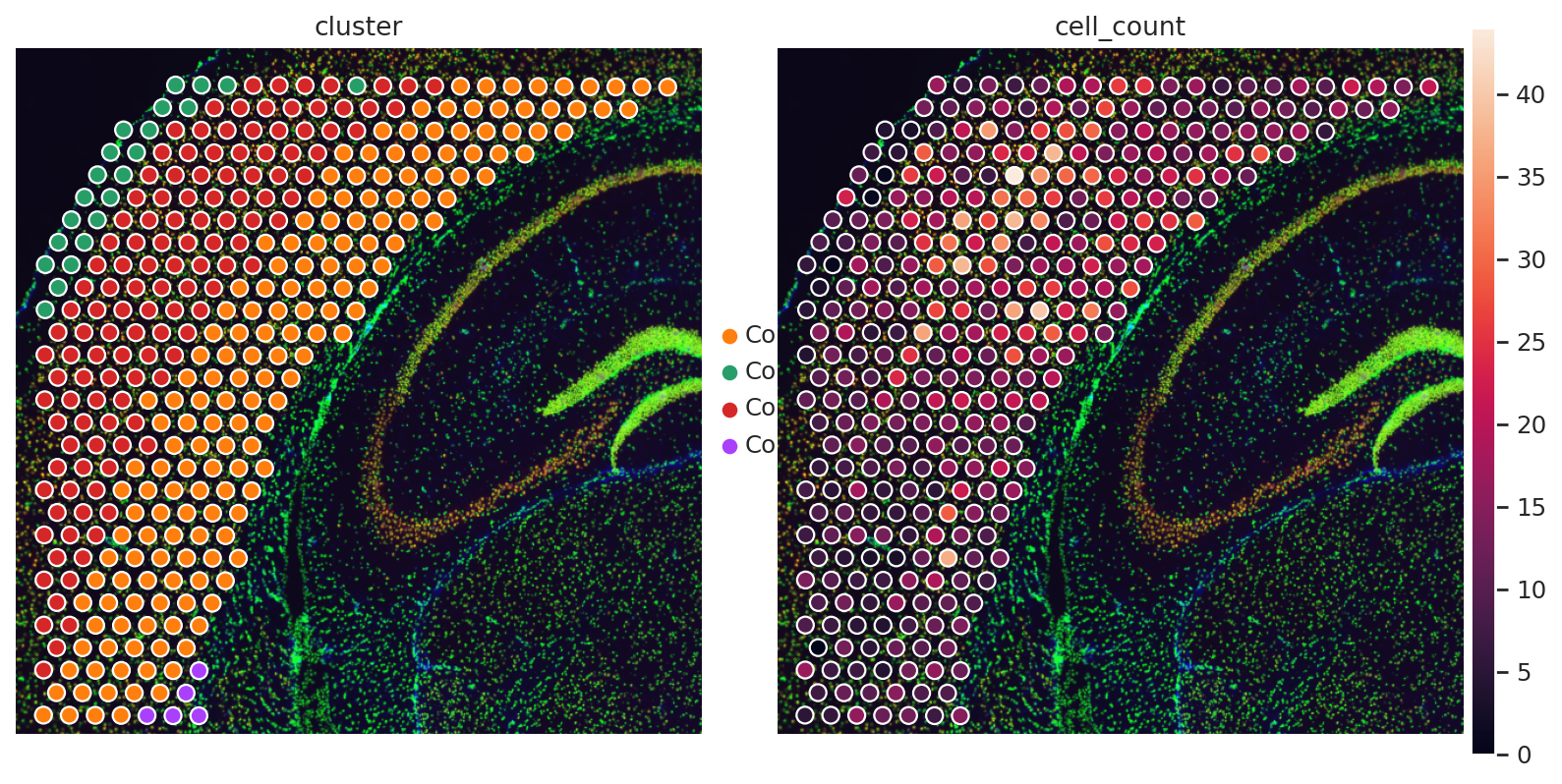
adata_sp.obs["cell_count"] = adata_sp.obsm["image_features"]["segmentation_label"]
sq.pl.spatial_scatter(adata_sp, color=["cluster", "cell_count"], frameon=False, wspace=0.01)
sc.tl.rank_genes_groups(adata_sc, groupby="cell_subclass", use_raw=False)
Find genes for mapping#
markers_df = pd.DataFrame(adata_sc.uns["rank_genes_groups"]["names"]).iloc[0:100, :]
genes_sc = np.unique(markers_df.melt().value.values)
genes_st = adata_sp.var_names.values
genes = list(set(genes_sc).intersection(set(genes_st)))
len(genes)
1280
Add training objects to mudata#
Here we compute all the density priors we need to run Tangram.
target_count = adata_sp.obs.cell_count.sum()
adata_sp.obs["density_prior"] = np.asarray(adata_sp.obs.cell_count) / target_count
rna_count_per_spot = np.asarray(adata_sp.X.sum(axis=1)).squeeze()
adata_sp.obs["rna_count_based_density"] = rna_count_per_spot / np.sum(rna_count_per_spot)
adata_sp.obs["uniform_density"] = np.ones(adata_sp.X.shape[0]) / adata_sp.X.shape[0]
adata_sp.var_names_make_unique()
mdata.mod["sp"].var_names_make_unique()
mdata.mod["sc"].var_names_make_unique()
mdata.mod["sp_train"] = mdata.mod["sp"][:, genes].copy()
mdata.mod["sc_train"] = mdata.mod["sc"][:, genes].copy()
# global
mdata.var_names = mdata.var_names.astype(str)
mdata.var_names_make_unique()
# per modality
for ad in mdata.mod.values():
ad.var_names = ad.var_names.astype(str)
ad.var_names_make_unique()
mdata.update()
Run Tangram#
Here we run the constrained mode; however, we could have set constrained=False, target_count=None, and density_prior_key="rna_count_based_density" to recover Tangram’s "cells" mode.
Tangram.setup_mudata(
mdata,
density_prior_key="density_prior",
modalities={
"density_prior_key": "sp_train",
"sc_layer": "sc_train",
"sp_layer": "sp_train",
},
)
model = Tangram(mdata, constrained=True, target_count=target_count)
model.train()
INFO Jax module moved to cuda:0.Note: Pytorch lightning will show GPU is not being used for the Trainer.
mapper = model.get_mapper_matrix()
mdata.mod["sc"].obsm["tangram_mapper"] = mapper
labels = mdata.mod["sc"].obs.cell_subclass
mdata.mod["sp"].obsm["tangram_ct_pred"] = model.project_cell_annotations(
mdata.mod["sc"], mdata.mod["sp"], mapper, labels
)
mdata.mod["sp_sc_projection"] = model.project_genes(mdata.mod["sc"], mdata.mod["sp"], mapper)
pred = adata_sp.obsm["tangram_ct_pred"]
if not isinstance(pred, pd.DataFrame):
pred = pd.DataFrame(pred, index=adata_sp.obs_names)
adata_sp.obs = pd.concat([adata_sp.obs, pred], axis=1)
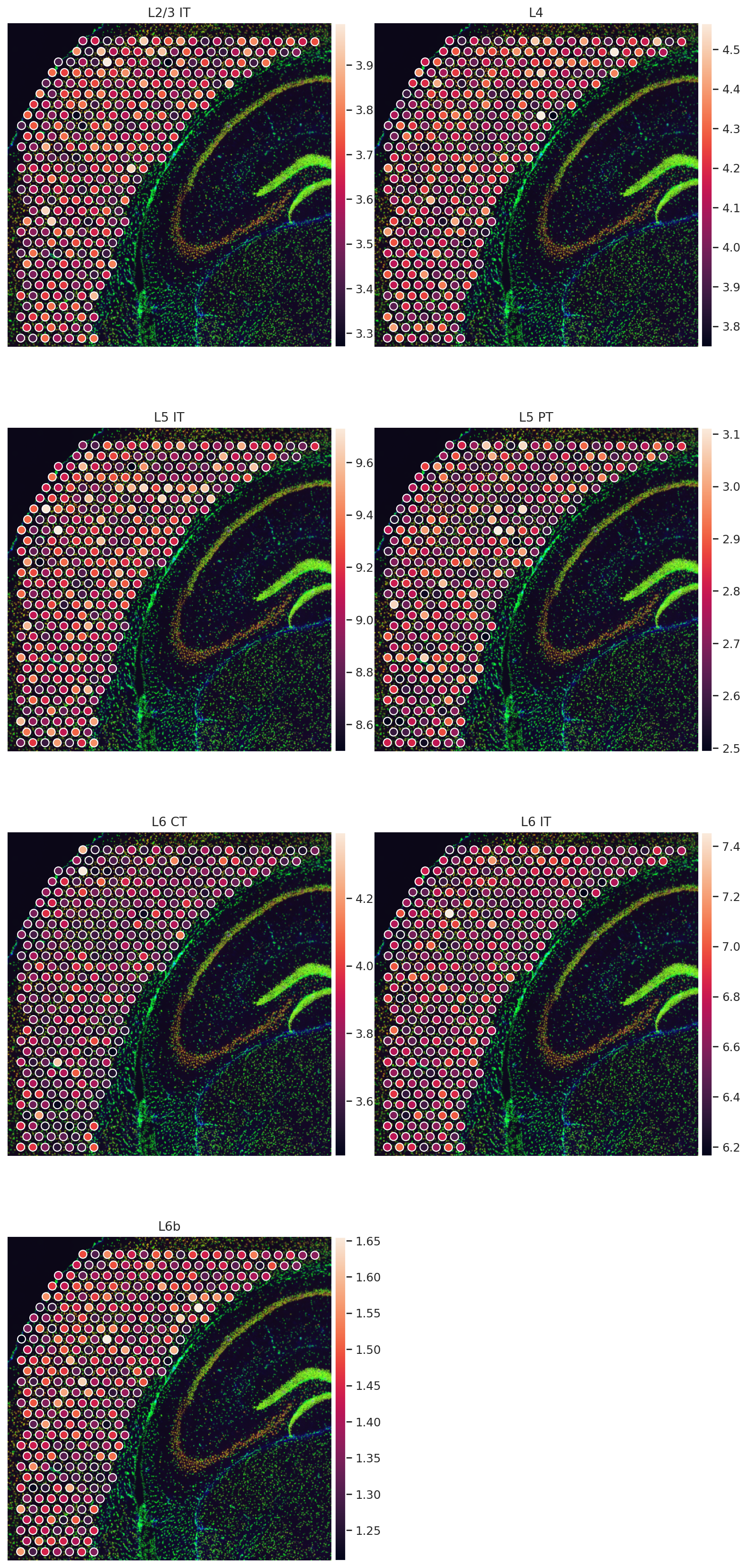
sq.pl.spatial_scatter(
adata_sp,
color=["L2/3 IT", "L4", "L5 IT", "L5 PT", "L6 CT", "L6 IT", "L6b"],
wspace=0.01,
ncols=2,
)