Decipher Quick Start Tutorial#
Decipher is a model for jointly analyzing single-cell RNA-seq samples from distinct conditions (e.g. normal vs perturbed samples). This tutorial will guide you through the steps to use Decipher to analyze a dataset.
Note: This tutorial currently only features a basic Decipher model. A more fleshed out implemention aligned with the original implementation is currently in development and will be released soon along with updates to this tutorial.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
[notice] A new release of pip is available: 24.3.1 -> 26.1.2
[notice] To update, run: pip install --upgrade pip
import os
import tempfile
import matplotlib.pyplot as plt
import scanpy as sc
import scvi
from scvi.external import Decipher
scvi.settings.seed = 0 # optional: ensures reproducibility
print("Last run with scvi-tools version:", scvi.__version__)
save_dir = tempfile.TemporaryDirectory()
Last run with scvi-tools version: 1.4.3
Preprocessing and model fitting#
For this tutorial, we will use a subset of the AML data from the Decipher preprint for the purpose of demonstrating how to use Decipher with scvi-tools.
adata_path = os.path.join(save_dir.name, "decipher_tutorial_data.h5ad")
adata = sc.read(
adata_path,
backup_url="https://exampledata.scverse.org/scvi-tools/data_decipher_tutorial.h5ad",
)
adata = adata[
~adata.obs["cell_type"].isin(["mep", "ery", "lympho"])
].copy() # subset to only include relevant cell types
adata
AnnData object with n_obs × n_vars = 16627 × 3130
obs: 'cell_type', 'origin', 'NPM1 mutation vs wild type'
uns: 'color_palette', 'decipher'
obsm: 'X_pca', 'X_tsne', 'decipher_v', 'decipher_z'
varm: 'PCs'
obsp: 'connectivities', 'distances'
Decipher does not require any additional covariates, and only optionally takes a layer indicating which layer of the AnnData object contains the raw count data.
Decipher.setup_anndata(adata)
Now we are ready to fit the model.
model = Decipher(adata)
model.train(
max_epochs=100,
batch_size=64,
early_stopping=True,
early_stopping_patience=10,
)
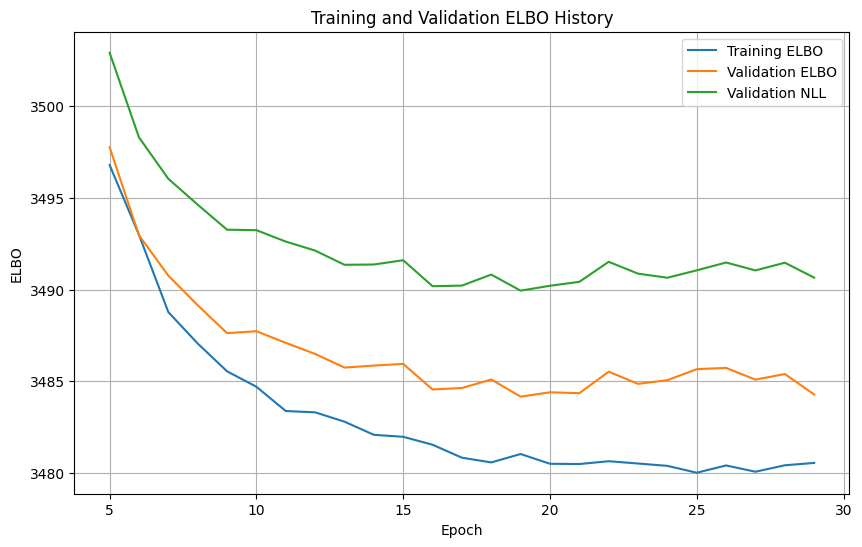
Monitored metric nll_validation did not improve in the last 10 records. Best score: 3489.938. Signaling Trainer to stop.
# save load functionality
save_path = f"./_decipher_models/test_notebook_model_{scvi.settings.seed}"
model.save(save_path, overwrite=True)
model = Decipher.load(save_path, adata)
INFO File ./_decipher_models/test_notebook_model_0/model.pt already downloaded
# Plot training and validation ELBO history
plt.figure(figsize=(10, 6))
plt.plot(model.history_["elbo_train"][5:], label="Training ELBO")
plt.plot(model.history_["elbo_validation"][5:], label="Validation ELBO")
plt.plot(model.history_["nll_validation"][5:], label="Validation NLL")
plt.xlabel("Epoch")
plt.ylabel("ELBO")
plt.title("Training and Validation ELBO History")
plt.legend()
plt.grid(True)
plt.show()
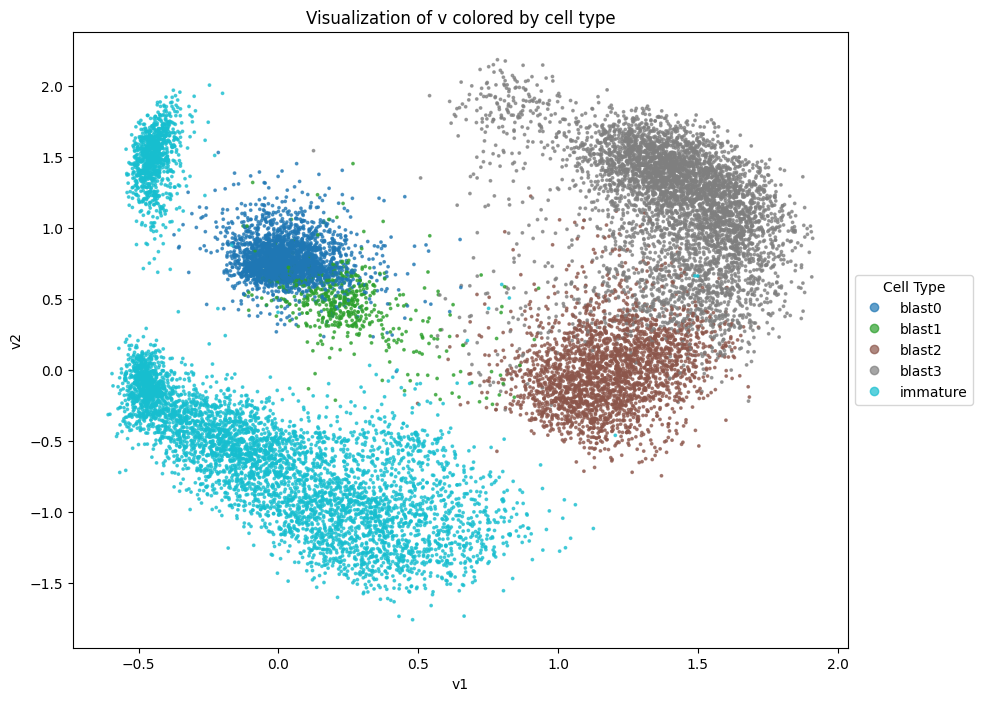
Visualize the latent representation#
Now that we have confirmed the model has converged, we can visualize the latent representation.
Notably, Decipher has two latent representations: v and z. v is a 2-dimensional latent representation which is amenable to direct visualization, while z is a higher-dimensional latent representation which is designed to capture more refined cell state information such as transitional intermediates.
v = model.get_latent_representation()
# Plot v and color by cell type
cell_type_mapping = adata.obs["cell_type"].astype("category").cat
plt.figure(figsize=(10, 8))
scatter = plt.scatter(v[:, 0], v[:, 1], c=cell_type_mapping.codes, cmap="tab10", alpha=0.7, s=3)
plt.legend(
scatter.legend_elements()[0],
cell_type_mapping.categories,
title="Cell Type",
loc="center left",
bbox_to_anchor=(1, 0.5),
)
plt.title("Visualization of v colored by cell type")
plt.xlabel("v1")
plt.ylabel("v2")
plt.show()
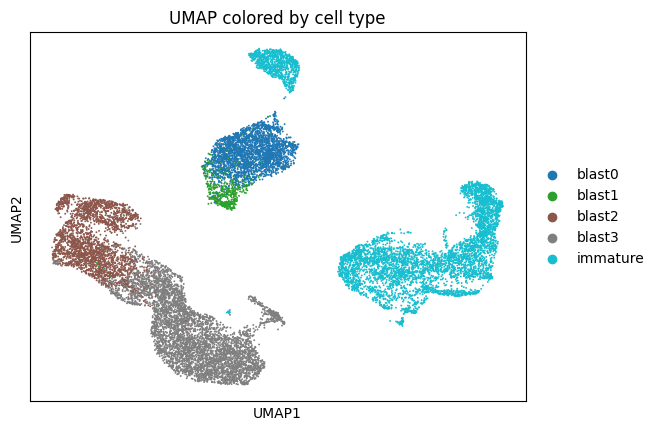
z = model.get_latent_representation(give_z=True)
adata.obsm["z"] = z
sc.pp.neighbors(adata, use_rep="z")
sc.tl.umap(adata, min_dist=0.3)
sc.pl.umap(
adata,
color=["cell_type"],
frameon=True,
palette="tab10",
ncols=1,
title="UMAP colored by cell type",
)