Train a scVI model using Anncollection dataloader wrapper#
In this tutorial we will show how to apply the annCollection wrapper in scvi-tools to load and train SCANVI model on several adata’s that are stored on disk
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import tempfile
from pathlib import Path
import anndata
import gdown
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import seaborn as sns
import torch
from anndata.experimental import AnnCollection
from scipy import sparse as sp
from scvi.dataloaders import CollectionAdapter
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.3.2
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
We will use 2 types of datasets : PBMC and Covid data, both from SCVI datasets repo
# the data is from this scvi reproducibility notebook
# https://yoseflab.github.io/scvi-tools-reproducibility/scarches_totalvi_seurat_data/
if Path("./pbmc_seurat_v4.h5ad").exists() and Path("./covid_cite.h5ad").exists():
print("Data already downloaded")
else:
gdown.download(
url="https://drive.google.com/uc?id=1X5N9rOaIqiGxZRyr1fyZ6NpDPeATXoaC",
output="pbmc_seurat_v4.h5ad",
quiet=False,
)
gdown.download(
url="https://drive.google.com/uc?id=1JgaXNwNeoEqX7zJL-jJD3cfXDGurMrq9",
output="covid_cite.h5ad",
quiet=False,
)
Data already downloaded
Preprocessing of the data#
covid = sc.read("covid_cite.h5ad")
pbmc = sc.read("pbmc_seurat_v4.h5ad")
pbmc.obs["dataset"] = "pbmc"
covid.obs["dataset"] = "covid"
# take annotations from the `pbmc` dataset and leave annotations in `covid` as an Unknown (test)
covid.obs["celltype.l1"] = "Unknown"
Note covid datasets has more genes than the pbmc. We manualy inersect the correct genes.
covid = covid[:, list(pbmc.var.index)]
# create a fake counts layer to test training
covid.layers["test"] = covid.X.copy()
pbmc.layers["test"] = pbmc.X.copy()
covid.raw = covid
pbmc.raw = pbmc
covid
AnnData object with n_obs × n_vars = 57669 × 20729
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'RNA_snn_res.0.4', 'seurat_clusters', 'set', 'Resp', 'disease', 'subj_code', 'covidpt_orhealth', 'mito', 'ncount', 'nfeat', 'bust_21', 'og_clust', 'severmod_other', 'og_clusts', 'nCount_ADT', 'nFeature_ADT', 'UMAP1', 'UMAP2', 'final_clust', 'final_clust_v2', 'new_pt_id', 'Resp_og', 'final_clust_withnum', 'final_clust_review', 'Age', 'Gender', 'Gender_num', 'dataset', 'celltype.l1'
obsm: 'pro_exp'
layers: 'test'
pbmc
AnnData object with n_obs × n_vars = 161764 × 20729
obs: 'nCount_ADT', 'nFeature_ADT', 'nCount_RNA', 'nFeature_RNA', 'orig.ident', 'lane', 'donor', 'time', 'celltype.l1', 'celltype.l2', 'celltype.l3', 'Phase', 'nCount_SCT', 'nFeature_SCT', 'X_index', 'dataset'
obsm: 'protein_counts'
layers: 'test'
# create an AnnCollection on a subset of the data
# we're subsetting purely for speed
adata = AnnCollection(
[covid, pbmc],
join_vars="inner",
join_obs="inner",
label="dataset",
)
adata
AnnCollection object with n_obs × n_vars = 219433 × 20729
constructed from 2 AnnData objects
view of layers: 'test'
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
collection_adapter = CollectionAdapter(adata)
collection_adapter
Adapter for:
AnnCollection object with n_obs × n_vars = 219433 × 20729
constructed from 2 AnnData objects
view of layers: 'test'
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
collection_adapter.adatas[0].X
<Compressed Sparse Row sparse matrix of dtype 'float32'
with 119919378 stored elements and shape (57669, 20729)>
But in this case we will run HVG selection first for both adatas together
# have an object of 2 adatas from the collection concatenated together
adatas = anndata.concat([collection_adapter.adatas[0], collection_adapter.adatas[1]])
we will do the usuall HVG selection and count transformation on the data
sc.pp.filter_genes(adatas, min_counts=3)
sc.pp.normalize_total(adatas, target_sum=1e4)
sc.pp.log1p(adatas)
sc.pp.highly_variable_genes(
adatas,
n_top_genes=1000,
subset=True,
layer="test",
flavor="seurat_v3",
batch_key="dataset",
)
adatas
AnnData object with n_obs × n_vars = 219433 × 1000
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
var: 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg'
layers: 'test'
We can now save the adatas to disk to be used in anncollection
adatas[adatas.obs.dataset == "covid"]
View of AnnData object with n_obs × n_vars = 57669 × 1000
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
var: 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'log1p', 'hvg'
layers: 'test'
adatas[adatas.obs.dataset == "pbmc"].write("pbmc_subset.h5ad")
adatas[adatas.obs.dataset == "covid"].write("covid_subset.h5ad")
Reload data after preprocessing into an AnndataCollection#
# we load the adataq in backed disk mode
covid_subset = sc.read("covid_subset.h5ad", backed="r")
pbmc_subset = sc.read("pbmc_subset.h5ad", backed="r")
Note that our count data is in a sparse form, which is the only one supported currently when using the AnnCollection Wrapper in SCVI-Tools
# create an AnnCollection on a subset of the adata's
adata = AnnCollection(
[covid_subset, pbmc_subset],
join_vars="inner",
join_obs="inner",
label="dataset",
)
print(adata)
AnnCollection object with n_obs × n_vars = 219433 × 1000
constructed from 2 AnnData objects
view of layers: 'test'
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
Build a wrapper AnnData around the collection#
collection_adapter = CollectionAdapter(adata)
collection_adapter
Adapter for:
AnnCollection object with n_obs × n_vars = 219433 × 1000
constructed from 2 AnnData objects
view of layers: 'test'
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nCount_ADT', 'nFeature_ADT', 'dataset', 'celltype.l1'
sp.issparse(collection_adapter.layers["test"])
True
scvi.model.SCANVI.setup_anndata(
collection_adapter,
layer="test",
batch_key="dataset",
labels_key="celltype.l1",
unlabeled_category="Unknown",
)
model = scvi.model.SCANVI(collection_adapter, n_latent=10)
# we're only training for a few epochs to show it works
model.train(max_epochs=25, early_stopping=True)
INFO Training for 5 epochs.
SCANVI_LATENT_KEY = "X_scanVI"
latent = model.get_latent_representation()
latent.shape
(219433, 10)
adatas.obsm[SCANVI_LATENT_KEY] = latent
Generate predictions that will include the covid unknown cells types
predictions = model.predict(collection_adapter)
adatas.obs["predictions_scanvi"] = predictions
adata.obs["predictions_scanvi"] = predictions
collection_adapter.obs["predictions_scanvi"] = predictions
Lets compare the PCA vs SCANVI Integrations UMAP results. In order to show the UMAP’s we will save the generated embeddings in the adatas object.
# run PCA then generate UMAP plots
sc.tl.pca(adatas)
sc.pp.neighbors(adatas, n_pcs=30, n_neighbors=20)
sc.tl.umap(adatas, min_dist=0.3)
sc.pl.umap(
adatas,
color=["predictions_scanvi", "dataset"],
frameon=False,
)
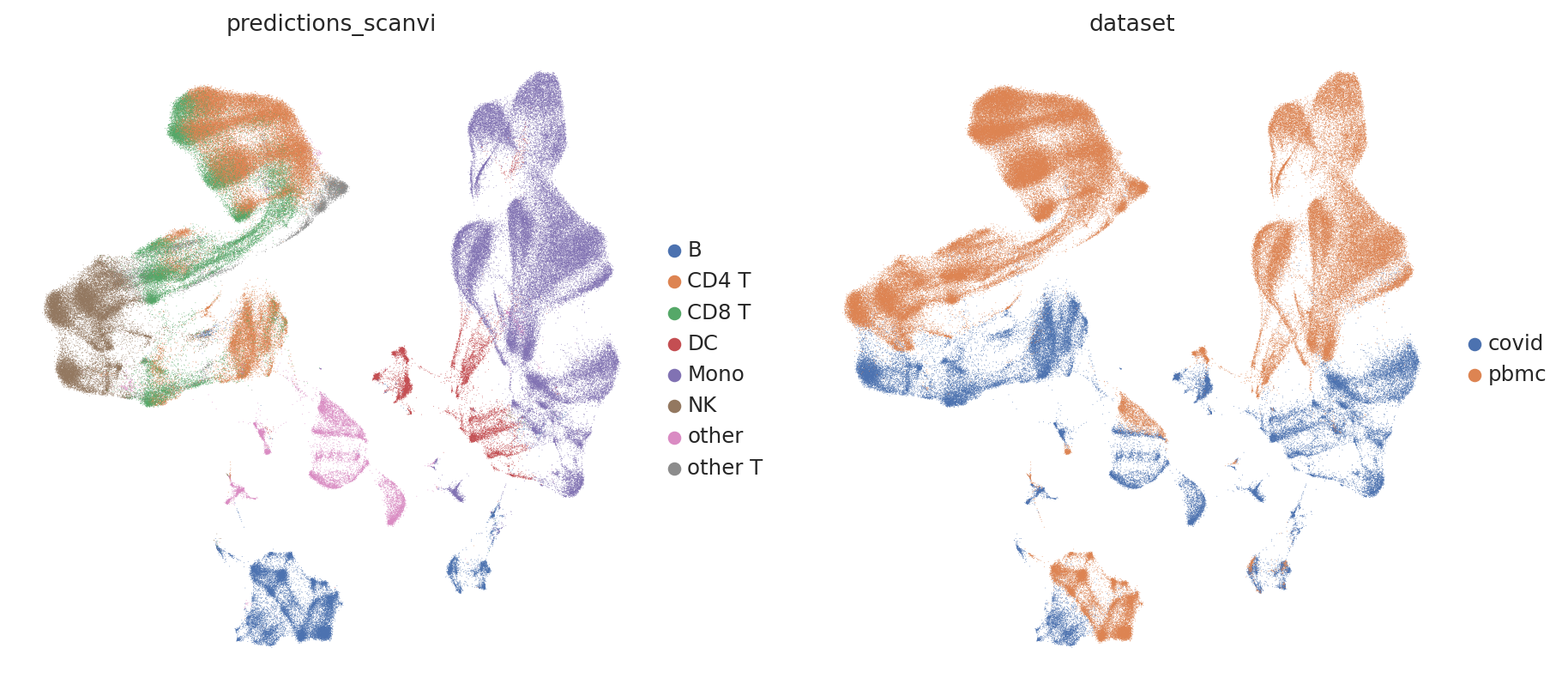
And for SCANVI Intgeration
# use scVI latent space for UMAP generation
sc.pp.neighbors(adatas, use_rep=SCANVI_LATENT_KEY)
sc.tl.umap(adatas, min_dist=0.3)
sc.pl.umap(
adatas,
color=["predictions_scanvi", "dataset"],
frameon=False,
)

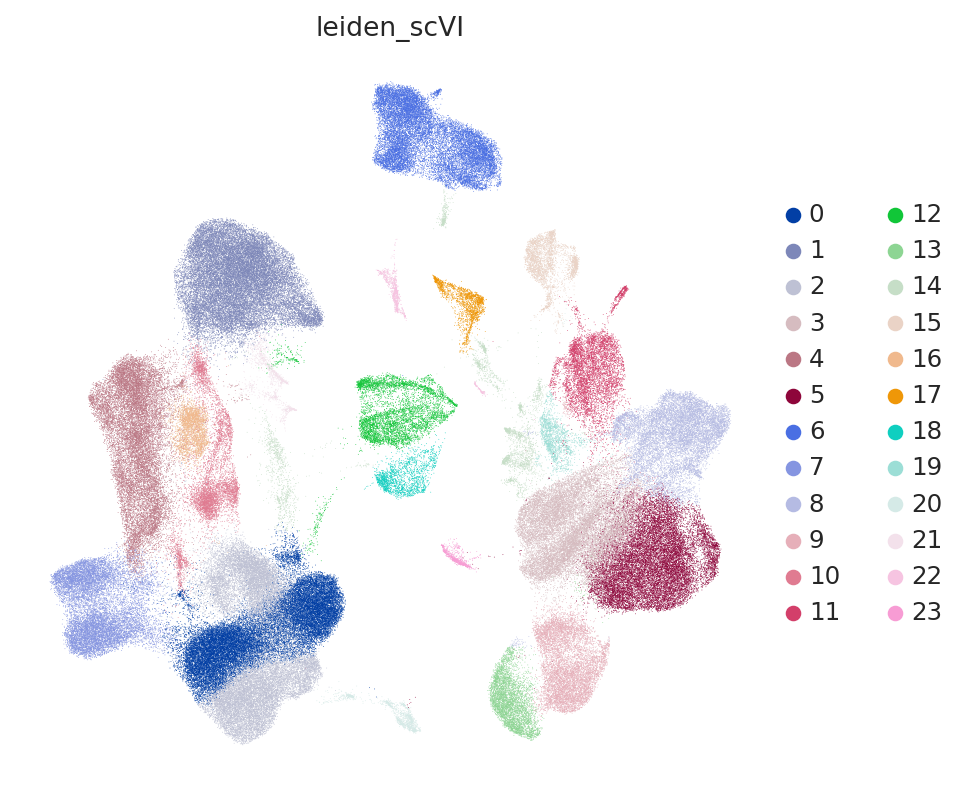
# neighbors were already computed using scVI
SCVI_CLUSTERS_KEY = "leiden_scVI"
sc.tl.leiden(adatas, key_added=SCVI_CLUSTERS_KEY, resolution=0.5)
sc.pl.umap(
adatas,
color=[SCVI_CLUSTERS_KEY],
frameon=False,
)
Confusion Matrix
df = adatas.obs.groupby(["celltype.l1", "predictions_scanvi"]).size().unstack(fill_value=0)
norm_df = df / df.sum(axis=0)
import matplotlib.pyplot as plt
plt.figure(figsize=(8, 8))
_ = plt.pcolor(norm_df)
_ = plt.xticks(np.arange(0.5, len(df.columns), 1), df.columns, rotation=90)
_ = plt.yticks(np.arange(0.5, len(df.index), 1), df.index)
plt.xlabel("Predicted")
plt.ylabel("Observed")
Text(0, 0.5, 'Observed')
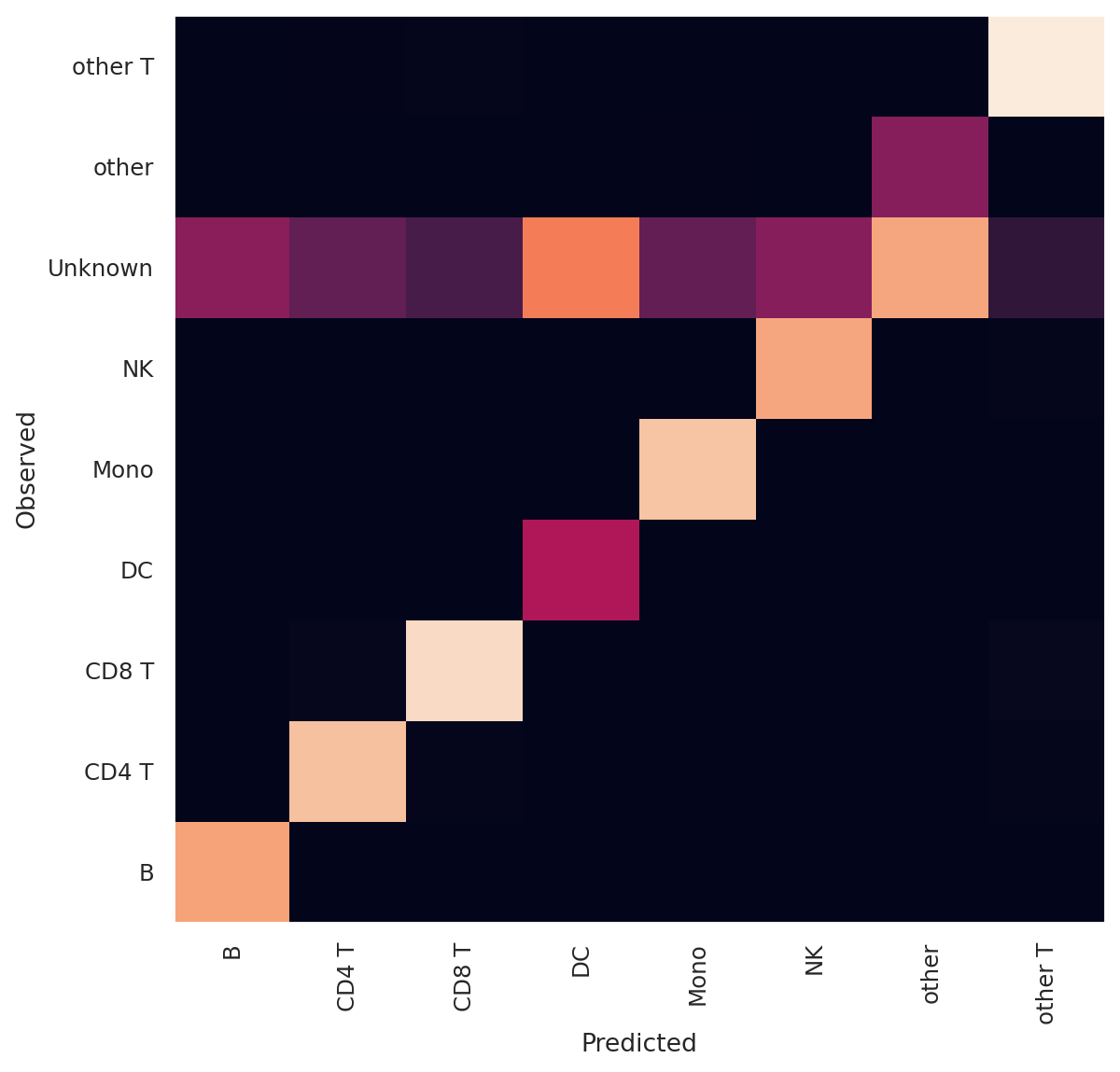
pd.crosstab(adatas.obs["celltype.l1"], adatas.obs["predictions_scanvi"])
| predictions_scanvi | B | CD4 T | CD8 T | DC | Mono | NK | other | other T |
|---|---|---|---|---|---|---|---|---|
| celltype.l1 | ||||||||
| B | 13794 | 0 | 1 | 0 | 3 | 1 | 1 | 0 |
| CD4 T | 2 | 40709 | 223 | 0 | 5 | 5 | 3 | 54 |
| CD8 T | 0 | 603 | 24741 | 0 | 1 | 19 | 2 | 103 |
| DC | 0 | 0 | 0 | 3488 | 99 | 0 | 2 | 0 |
| Mono | 1 | 2 | 1 | 19 | 48975 | 0 | 12 | 0 |
| NK | 0 | 13 | 52 | 0 | 0 | 18528 | 2 | 69 |
| Unknown | 5850 | 11506 | 4878 | 5865 | 14061 | 7577 | 7150 | 782 |
| other | 2 | 79 | 24 | 2 | 376 | 16 | 2940 | 3 |
| other T | 0 | 278 | 219 | 0 | 2 | 59 | 0 | 6231 |
Compare results#
from scib_metrics.benchmark import Benchmarker
bm = Benchmarker(
adatas[list(np.random.choice(np.arange(adatas.n_obs), size=10000, replace=False)), :],
batch_key="dataset",
label_key="predictions_scanvi",
embedding_obsm_keys=["X_pca", "X_scanVI"],
n_jobs=-1,
)
bm.benchmark()
INFO 8 clusters consist of a single batch or are too small. Skip.
INFO 8 clusters consist of a single batch or are too small. Skip.
bm.plot_results_table(min_max_scale=False)
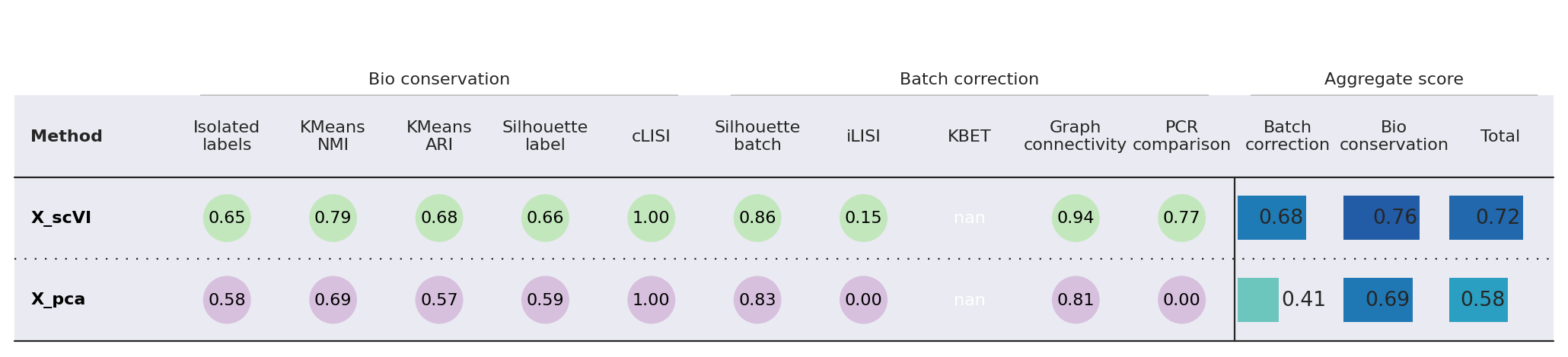
<plottable.table.Table at 0x7bd284b92990>
Save and load model
model.save("model_scanvi_anncollection", save_anndata=False, overwrite=True)
# Load model again
loaded_model = scvi.model.SCANVI.load("model_scanvi_anncollection", adata=collection_adapter)
loaded_model
INFO File model_scanvi_anncollection/model.pt already downloaded
ScanVI Model with the following params: unlabeled_category: Unknown, n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: zinb Training status: Trained Model's adata is minified?: False
# We can continue training the loaded model
loaded_model.train(max_epochs=1)
INFO Training for 1 epochs.
# loaded_model.registry