Use pretrained models of scVI-hub for Tahoe100M#
This notebook represent an example of how to perform downstream anaylsis on a pretrained SCVI model that was minified and saved in a model hub. In this case we use the model that was trained based on the Tahoe100M dataset , by Vevo Therapuetics. See link to model hub here
Steps performed:
Loading the minified data from AWS
Setting up minified model with minified data
Visualize the latent space
Perform differential expression and visualize with interactive volcano plot and heatmap using Plotly
import tempfile
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import scvi.hub
import seaborn as sns
import torch
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.3.2
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
Get the data#
We start by downloading the model from its hub. Note that the model is very large therefore it will take time to being download.
tahoe_hubmodel = scvi.hub.HubModel.pull_from_huggingface_hub(
repo_name="vevotx/Tahoe-100M-SCVI-v1", cache_dir="."
)
We can see the model card
# This will be a bit long to scroll, but has all the info
# tahoe_hubmodel
We will extract he model and the minifed adata
tahoe = tahoe_hubmodel.model
tahoe
INFO Loading model...
INFO File ./models--vevotx--Tahoe-100M-SCVI-v1/snapshots/b5283a73fbbed812a95264ace360da538b20af89/model.pt
already downloaded
SCVI model with the following parameters: n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: nb, latent_distribution: normal. Training status: Trained Model's adata is minified?: True
tahoe.adata
AnnData object with n_obs × n_vars = 95624334 × 62710
obs: 'sample', 'species', 'gene_count', 'tscp_count', 'mread_count', 'bc1_wind', 'bc2_wind', 'bc3_wind', 'bc1_well', 'bc2_well', 'bc3_well', 'id', 'drugname_drugconc', 'drug', 'INT_ID', 'NUM.SNPS', 'NUM.READS', 'demuxlet_call', 'BEST.LLK', 'NEXT.LLK', 'DIFF.LLK.BEST.NEXT', 'BEST.POSTERIOR', 'SNG.POSTERIOR', 'SNG.BEST.LLK', 'SNG.NEXT.LLK', 'SNG.ONLY.POSTERIOR', 'DBL.BEST.LLK', 'DIFF.LLK.SNG.DBL', 'sublibrary', 'BARCODE', 'pcnt_mito', 'S_score', 'G2M_score', 'phase', 'pass_filter', 'dataset', '_scvi_batch', '_scvi_labels', '_scvi_observed_lib_size', 'plate', 'Cell_Name_Vevo', 'Cell_ID_Cellosaur', 'observed_lib_size'
var: 'gene_id', 'genome', 'SUB_LIB_ID'
uns: '_scvi_adata_minify_type', '_scvi_manager_uuid', '_scvi_uuid'
obsm: 'X_latent_qzm', 'X_latent_qzv', 'scvi_latent_qzm', 'scvi_latent_qzv'
layers: 'counts'
tahoe.view_anndata_setup()
Anndata setup with scvi-tools version 1.3.2.
Setup via `SCVI.setup_anndata` with arguments:
{ │ 'layer': 'counts', │ 'batch_key': None, │ 'labels_key': None, │ 'size_factor_key': 'tscp_count', │ 'categorical_covariate_keys': None, │ 'continuous_covariate_keys': None }
Summary Statistics ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━┓ ┃ Summary Stat Key ┃ Value ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━┩ │ n_batch │ 1 │ │ n_cells │ 95624334 │ │ n_extra_categorical_covs │ 0 │ │ n_extra_continuous_covs │ 0 │ │ n_labels │ 1 │ │ n_latent_qzm │ 10 │ │ n_latent_qzv │ 10 │ │ n_vars │ 62710 │ └──────────────────────────┴──────────┘
Data Registry ┏━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ┃ Registry Key ┃ scvi-tools Location ┃ ┡━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ │ X │ adata.layers['counts'] │ │ batch │ adata.obs['_scvi_batch'] │ │ labels │ adata.obs['_scvi_labels'] │ │ latent_qzm │ adata.obsm['scvi_latent_qzm'] │ │ latent_qzv │ adata.obsm['scvi_latent_qzv'] │ │ minify_type │ adata.uns['_scvi_adata_minify_type'] │ │ observed_lib_size │ adata.obs['observed_lib_size'] │ │ size_factor │ adata.obs['tscp_count'] │ └───────────────────┴──────────────────────────────────────┘
batch State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['_scvi_batch'] │ 0 │ 0 │ └──────────────────────────┴────────────┴─────────────────────┘
labels State Registry ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━┓ ┃ Source Location ┃ Categories ┃ scvi-tools Encoding ┃ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━┩ │ adata.obs['_scvi_labels'] │ 0 │ 0 │ └───────────────────────────┴────────────┴─────────────────────┘
# from pprint import pprint
# pprint(tahoe.registry)
Get the latent space#
SCVI_LATENT_KEY = "X_scVI"
latent = tahoe.get_latent_representation()
tahoe.adata.obsm[SCVI_LATENT_KEY] = latent
latent.shape
(95624334, 10)
# we visualize a subset of the results to avoid meaningless overplotting.
subset_adata = tahoe.adata[np.random.randint(0, tahoe.adata.shape[0], (1_000_000,))]
subset_adata
View of AnnData object with n_obs × n_vars = 1000000 × 62710
obs: 'sample', 'species', 'gene_count', 'tscp_count', 'mread_count', 'bc1_wind', 'bc2_wind', 'bc3_wind', 'bc1_well', 'bc2_well', 'bc3_well', 'id', 'drugname_drugconc', 'drug', 'INT_ID', 'NUM.SNPS', 'NUM.READS', 'demuxlet_call', 'BEST.LLK', 'NEXT.LLK', 'DIFF.LLK.BEST.NEXT', 'BEST.POSTERIOR', 'SNG.POSTERIOR', 'SNG.BEST.LLK', 'SNG.NEXT.LLK', 'SNG.ONLY.POSTERIOR', 'DBL.BEST.LLK', 'DIFF.LLK.SNG.DBL', 'sublibrary', 'BARCODE', 'pcnt_mito', 'S_score', 'G2M_score', 'phase', 'pass_filter', 'dataset', '_scvi_batch', '_scvi_labels', '_scvi_observed_lib_size', 'plate', 'Cell_Name_Vevo', 'Cell_ID_Cellosaur', 'observed_lib_size'
var: 'gene_id', 'genome', 'SUB_LIB_ID'
uns: '_scvi_adata_minify_type', '_scvi_manager_uuid', '_scvi_uuid'
obsm: 'X_latent_qzm', 'X_latent_qzv', 'scvi_latent_qzm', 'scvi_latent_qzv', 'X_scVI'
layers: 'counts'
subset_adata.X.shape
(1000000, 62710)
import gc
gc.collect()
0
Exploritory Analysis#
tahoe.adata.obs.head()
| sample | species | gene_count | tscp_count | mread_count | bc1_wind | bc2_wind | bc3_wind | bc1_well | bc2_well | ... | phase | pass_filter | dataset | _scvi_batch | _scvi_labels | _scvi_observed_lib_size | plate | Cell_Name_Vevo | Cell_ID_Cellosaur | observed_lib_size | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BARCODE_SUB_LIB_ID | |||||||||||||||||||||
| 01_001_052-lib_1105 | smp_1783 | hg38 | 1878 | 2893 | 3284 | 1 | 1 | 52 | A1 | A1 | ... | G2M | full | 0 | 0 | 0 | 2893 | 4 | PANC-1 | CVCL_0480 | 2893 |
| 01_001_105-lib_1105 | smp_1783 | hg38 | 1765 | 2434 | 2764 | 1 | 1 | 105 | A1 | A1 | ... | G2M | full | 0 | 0 | 0 | 2434 | 4 | SW480 | CVCL_0546 | 2434 |
| 01_001_165-lib_1105 | smp_1783 | hg38 | 3174 | 5691 | 6454 | 1 | 1 | 165 | A1 | A1 | ... | G2M | full | 0 | 0 | 0 | 5691 | 4 | SW1417 | CVCL_1717 | 5691 |
| 01_003_094-lib_1105 | smp_1783 | hg38 | 1380 | 1804 | 2050 | 1 | 3 | 94 | A1 | A3 | ... | G2M | full | 0 | 0 | 0 | 1804 | 4 | SW1417 | CVCL_1717 | 1804 |
| 01_003_164-lib_1105 | smp_1783 | hg38 | 1179 | 1514 | 1715 | 1 | 3 | 164 | A1 | A3 | ... | G1 | full | 0 | 0 | 0 | 1514 | 4 | A498 | CVCL_1056 | 1514 |
5 rows × 43 columns
# confuison table of plates and cell type (they are spread evently)
pd.crosstab(subset_adata.obs.plate, subset_adata.obs.Cell_ID_Cellosaur)
| Cell_ID_Cellosaur | CVCL_0023 | CVCL_0332 | CVCL_0292 | CVCL_1285 | CVCL_0069 | CVCL_1056 | CVCL_1381 | CVCL_0218 | CVCL_0320 | CVCL_0546 | ... | CVCL_0371 | CVCL_0099 | CVCL_0028 | CVCL_1550 | CVCL_0131 | CVCL_C466 | CVCL_1547 | CVCL_1495 | CVCL_1531 | CVCL_1517 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| plate | |||||||||||||||||||||
| 1 | 1605 | 866 | 785 | 1649 | 699 | 1172 | 1128 | 1056 | 1648 | 3410 | ... | 1543 | 473 | 285 | 1427 | 1423 | 1406 | 1049 | 905 | 23 | 1771 |
| 2 | 2331 | 1249 | 1288 | 2660 | 1063 | 1856 | 1624 | 1344 | 2035 | 5109 | ... | 2093 | 679 | 494 | 1970 | 2240 | 1911 | 1606 | 1311 | 25 | 2282 |
| 3 | 1286 | 817 | 475 | 1533 | 538 | 1123 | 1031 | 537 | 1131 | 2394 | ... | 1341 | 326 | 241 | 1520 | 1559 | 785 | 903 | 716 | 21 | 1570 |
| 4 | 2181 | 1157 | 1044 | 2310 | 1093 | 1867 | 1616 | 1089 | 1681 | 4242 | ... | 1741 | 645 | 530 | 1734 | 2014 | 1361 | 1612 | 1274 | 30 | 1752 |
| 5 | 1931 | 1209 | 684 | 2282 | 1056 | 1975 | 1560 | 786 | 1463 | 3108 | ... | 1361 | 532 | 546 | 1690 | 2210 | 1157 | 1417 | 1194 | 36 | 1727 |
| 6 | 2321 | 1122 | 1261 | 2508 | 1307 | 1935 | 1811 | 1153 | 1689 | 4736 | ... | 1774 | 814 | 759 | 1763 | 2212 | 1754 | 1541 | 1364 | 26 | 1748 |
| 7 | 1379 | 1147 | 796 | 2001 | 968 | 1843 | 1492 | 693 | 1149 | 3210 | ... | 1176 | 800 | 246 | 1408 | 1015 | 755 | 942 | 1200 | 10 | 764 |
| 8 | 2063 | 1550 | 1696 | 2856 | 1642 | 2380 | 2062 | 1472 | 1898 | 6383 | ... | 2122 | 1664 | 443 | 1886 | 1592 | 1171 | 1460 | 1885 | 20 | 1018 |
| 9 | 1340 | 1084 | 1019 | 1896 | 967 | 1702 | 1322 | 924 | 1348 | 4024 | ... | 1415 | 862 | 294 | 1291 | 1112 | 793 | 900 | 1212 | 13 | 846 |
| 10 | 1945 | 1537 | 1342 | 2968 | 1433 | 2434 | 1897 | 1241 | 1732 | 5459 | ... | 1712 | 1232 | 396 | 1647 | 1474 | 1201 | 1340 | 1657 | 19 | 1143 |
| 11 | 1843 | 1716 | 1122 | 2866 | 1097 | 2602 | 1616 | 1083 | 1671 | 4643 | ... | 1593 | 991 | 248 | 1462 | 1464 | 1302 | 1281 | 1596 | 14 | 1188 |
| 12 | 2616 | 1963 | 1863 | 3943 | 1876 | 3353 | 2500 | 1444 | 2108 | 7034 | ... | 2194 | 1846 | 621 | 2051 | 1981 | 1644 | 1710 | 2084 | 16 | 1296 |
| 13 | 2026 | 1555 | 1568 | 2931 | 1638 | 2619 | 2005 | 1245 | 1728 | 5560 | ... | 1844 | 1444 | 410 | 1937 | 1610 | 1156 | 1324 | 1784 | 21 | 1024 |
| 14 | 1814 | 1028 | 887 | 2220 | 939 | 1808 | 1274 | 1170 | 1604 | 3967 | ... | 1618 | 528 | 371 | 1331 | 2011 | 1339 | 1338 | 1124 | 23 | 1901 |
14 rows × 50 columns
# integration of drugs and cell type (certain type of cancer are given certian type of drugs)
df = subset_adata.obs.groupby(["drug", "Cell_ID_Cellosaur"]).size().unstack(fill_value=0)
norm_df = df / df.sum(axis=0)
plt.figure(figsize=(8, 8))
_ = plt.pcolor(norm_df)
_ = plt.xticks(np.arange(0.5, len(df.columns), 1), df.columns, rotation=90)
_ = plt.yticks(np.arange(0.5, len(df.index), 1), df.index)
plt.xlabel("Cell_ID_Cellosaur")
plt.ylabel("drug")
Text(0, 0.5, 'drug')

Visualization with batch correction (scVI)#
gc.collect()
0
# use scVI latent space for UMAP generation
sc.pp.neighbors(subset_adata, use_rep=SCVI_LATENT_KEY)
sc.tl.umap(subset_adata, min_dist=0.3)
# because we have so many drugs, we wont plot the umap here
# sc.pl.umap(
# subset_adata,
# color=["drug"],
# frameon=False,
# )
sc.pl.umap(
subset_adata,
color=["plate", "Cell_ID_Cellosaur"],
ncols=2,
frameon=False,
)
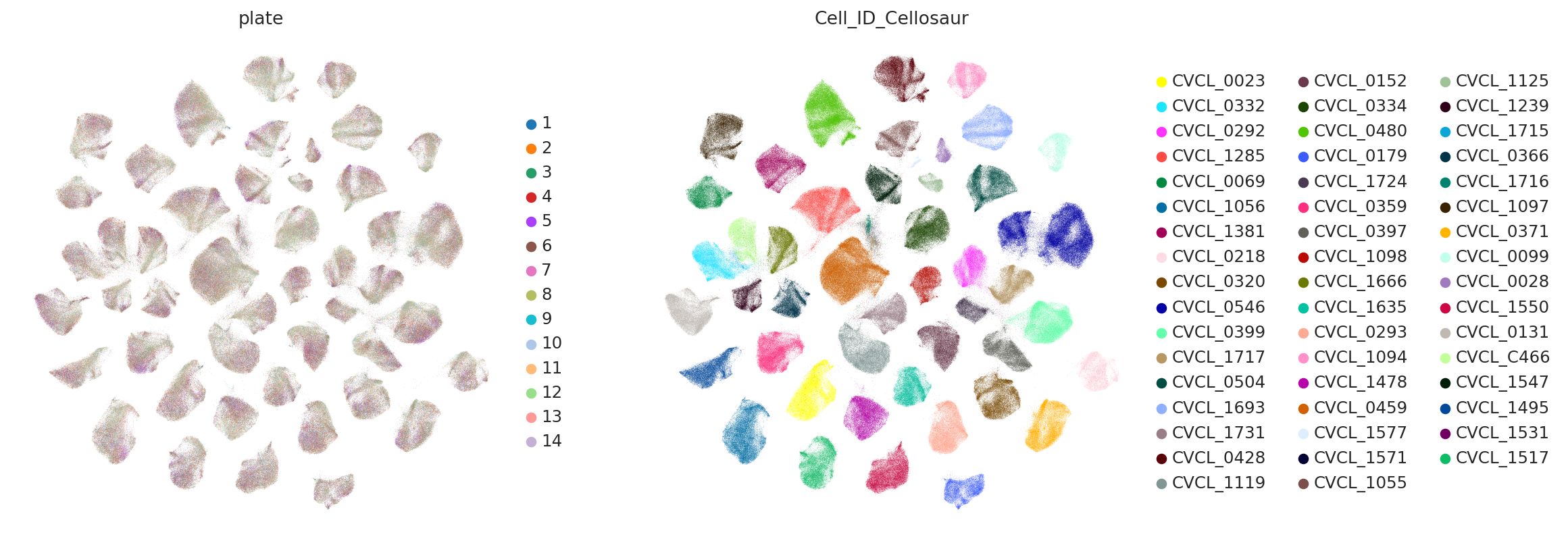
Visualization PCA#
In order to save time we will subset the data even further
subset_adata2 = subset_adata[np.random.randint(0, subset_adata.shape[0], (100_000,))]
subset_adata2
View of AnnData object with n_obs × n_vars = 100000 × 62710
obs: 'sample', 'species', 'gene_count', 'tscp_count', 'mread_count', 'bc1_wind', 'bc2_wind', 'bc3_wind', 'bc1_well', 'bc2_well', 'bc3_well', 'id', 'drugname_drugconc', 'drug', 'INT_ID', 'NUM.SNPS', 'NUM.READS', 'demuxlet_call', 'BEST.LLK', 'NEXT.LLK', 'DIFF.LLK.BEST.NEXT', 'BEST.POSTERIOR', 'SNG.POSTERIOR', 'SNG.BEST.LLK', 'SNG.NEXT.LLK', 'SNG.ONLY.POSTERIOR', 'DBL.BEST.LLK', 'DIFF.LLK.SNG.DBL', 'sublibrary', 'BARCODE', 'pcnt_mito', 'S_score', 'G2M_score', 'phase', 'pass_filter', 'dataset', '_scvi_batch', '_scvi_labels', '_scvi_observed_lib_size', 'plate', 'Cell_Name_Vevo', 'Cell_ID_Cellosaur', 'observed_lib_size'
var: 'gene_id', 'genome', 'SUB_LIB_ID'
uns: '_scvi_adata_minify_type', '_scvi_manager_uuid', '_scvi_uuid', 'neighbors', 'umap', 'plate_colors', 'Cell_ID_Cellosaur_colors'
obsm: 'X_latent_qzm', 'X_latent_qzv', 'scvi_latent_qzm', 'scvi_latent_qzv', 'X_scVI', 'X_umap'
layers: 'counts'
obsp: 'distances', 'connectivities'
# run PCA then generate UMAP plots (here no results will be shown as no raw data exists)
sc.tl.pca(subset_adata2)
sc.pp.neighbors(subset_adata2, n_pcs=50, n_neighbors=50)
sc.tl.umap(subset_adata2, min_dist=0.1)
# sc.pl.umap(
# subset_adata2,
# color=["plate", "Cell_ID_Cellosaur"],
# frameon=False,
# )
Clustering on the scVI latent space#
# neighbors were already computed using scVI
SCVI_CLUSTERS_KEY = "leiden_scVI"
sc.tl.leiden(subset_adata, key_added=SCVI_CLUSTERS_KEY, resolution=0.5)
sc.pl.umap(
subset_adata,
color=[SCVI_CLUSTERS_KEY],
frameon=False,
)

# integration of leiden clsuters and cell type
df = subset_adata.obs.groupby(["leiden_scVI", "Cell_ID_Cellosaur"]).size().unstack(fill_value=0)
norm_df = df / df.sum(axis=0)
plt.figure(figsize=(8, 8))
_ = plt.pcolor(norm_df)
_ = plt.xticks(np.arange(0.5, len(df.columns), 1), df.columns, rotation=90)
_ = plt.yticks(np.arange(0.5, len(df.index), 1), df.index)
plt.xlabel("Cell_ID_Cellosaur")
plt.ylabel("leiden_scVI")
Text(0, 0.5, 'leiden_scVI')

SCANVI#
Running scanvi from the scvi model will require the original counts matrix and cant be done, as count matrix is all 0.
gc.collect()
0
tahoe.save("tahoe_tmp", save_anndata=True, overwrite=True)
tahoe_loaded = scvi.model.SCVI.load("tahoe_tmp", adata=tahoe.adata)
INFO File tahoe_tmp/model.pt already downloaded
tahoe_loaded.minify_adata(minified_data_type="latent_posterior_parameters_with_counts")
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
INFO Generating sequential column names
INFO Generating sequential column names
# predict on drug?
tahoe_scanvi = scvi.model.SCANVI.from_scvi_model(
tahoe_loaded,
unlabeled_category="Unknown",
labels_key="drug",
)
tahoe_loaded.adata.X
<Compressed Sparse Row sparse matrix of dtype 'float64'
with 0 stored elements and shape (95624334, 62710)>
# tahoe_scanvi.train(max_epochs=5,batch_size=64) #this fails in the workstation
predictions = tahoe_scanvi.predict(subset_adata)
subset_adata.obs["predictions_scanvi"] = predictions
# predictions
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
See the predictions confusion matrix
df = subset_adata.obs.groupby(["drug", "predictions_scanvi"]).size().unstack(fill_value=0)
norm_df = df / df.sum(axis=0)
print("The overall Accuracy is:", np.round(np.trace(norm_df), 2))
The overall Accuracy is: 0.63
We create the scanvi embeddings as well (if model was trained)
SCANVI_LATENT_KEY = "X_scanVI"
# latent_scanvi = tahoe_scanvi.get_latent_representation(subset_adata2)
# subset_adata2.obsm[SCANVI_LATENT_KEY] = latent_scanvi
# use scVI latent space for UMAP generation
# sc.pp.neighbors(subset_adata2, use_rep=SCANVI_LATENT_KEY)
# sc.tl.umap(subset_adata2, min_dist=0.3)
# sc.pl.umap(
# subset_adata2,
# color=["plate", "Cell_ID_Cellosaur"],
# ncols=2,
# frameon=False,
# )
Perform Integration Analysis#
from scib_metrics.benchmark import Benchmarker
bm = Benchmarker(
subset_adata2,
batch_key="plate",
label_key="Cell_ID_Cellosaur",
embedding_obsm_keys=["X_pca", "X_scVI"],
n_jobs=-1,
)
bm.benchmark()
INFO 50 clusters consist of a single batch or are too small. Skip.
INFO 50 clusters consist of a single batch or are too small. Skip.
bm.plot_results_table(min_max_scale=False)

<plottable.table.Table at 0x7f036c17d670>
Performing Differential Expression in scVI#
While we only have access to the minified data, we can still perform downstream analysis using the generative part of the model. For example here, we will do it on a cluster of DMSO_TF controls vs the drug Harringtonine that is used for protein synthesis inhibitor per the cell line CVCL_0459 which is typicaly associated with Lung large cell carcinoma, a sub type of NSCLC. We also choose to use the sub group of G2M cell cycle phase.
tahoe.adata.obs["drug"].value_counts().head()
drug
DMSO_TF 2205786
Adagrasib 1433284
Afatinib 696323
Almonertinib (mesylate) 592031
Cytarabine 551715
Name: count, dtype: int64
tahoe.adata.obs["Cell_ID_Cellosaur"].value_counts().head()
Cell_ID_Cellosaur
CVCL_0546 6040371
CVCL_0459 5736238
CVCL_0480 4089586
CVCL_1285 3307302
CVCL_0399 3013246
Name: count, dtype: int64
scVI provides several options to identify the two populations of interest.
cell_line = "CVCL_0459"
cell_cycle = "G2M"
drug1 = "Harringtonine"
cell_idx1 = np.logical_and(
tahoe.adata.obs["Cell_ID_Cellosaur"] == cell_line,
tahoe.adata.obs["drug"] == drug1,
tahoe.adata.obs["phase"] == cell_cycle,
)
print(sum(cell_idx1), "cells from drug", drug1)
drug2 = "DMSO_TF"
cell_idx2 = np.logical_and(
tahoe.adata.obs["Cell_ID_Cellosaur"] == cell_line,
tahoe.adata.obs["drug"] == drug2,
tahoe.adata.obs["phase"] == cell_cycle,
)
print(sum(cell_idx2), "cells of drug", drug2)
17297 cells from drug Harringtonine
126345 cells of drug DMSO_TF
A simple DE analysis can then be performed using the following command
de_change = tahoe.differential_expression(
idx1=cell_idx1,
idx2=cell_idx2,
all_stats=True,
batch_correction=True,
mode="change",
delta=0.05,
pseudocounts=1e-12,
)
Volcano plot with p-values
de_change["log10_pscore"] = np.log10(de_change["proba_not_de"] + 1e-6)
de_change = de_change.join(tahoe.adata.var, how="inner")
de_change = de_change.loc[np.max(de_change[["scale1", "scale2"]], axis=1) > 1e-4]
de_change.head()
| proba_de | proba_not_de | bayes_factor | scale1 | scale2 | pseudocounts | delta | lfc_mean | lfc_median | lfc_std | ... | raw_mean2 | non_zeros_proportion1 | non_zeros_proportion2 | raw_normalized_mean1 | raw_normalized_mean2 | is_de_fdr_0.05 | log10_pscore | gene_id | genome | SUB_LIB_ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_name | |||||||||||||||||||||
| FAM172A | 0.9922 | 0.0078 | -4.845800 | 0.000152 | 0.000431 | 1.000000e-12 | 0.05 | -1.552615 | -1.596409 | 0.511571 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | True | -2.107850 | ENSG00000113391 | hg38 | lib_1105 |
| CXCL3 | 0.9922 | 0.0078 | 4.845800 | 0.001353 | 0.000023 | 1.000000e-12 | 0.05 | 5.886231 | 6.114166 | 1.914412 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | True | -2.107850 | ENSG00000163734 | hg38 | lib_1105 |
| KLF10 | 0.9920 | 0.0080 | 4.820280 | 0.001278 | 0.000072 | 1.000000e-12 | 0.05 | 3.911682 | 4.109592 | 1.268676 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | True | -2.096856 | ENSG00000155090 | hg38 | lib_1105 |
| PIM3 | 0.9914 | 0.0086 | 4.747355 | 0.000254 | 0.000021 | 1.000000e-12 | 0.05 | 3.689933 | 3.854218 | 1.218505 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | True | -2.065451 | ENSG00000198355 | hg38 | lib_1105 |
| NFKBIB | 0.9906 | 0.0094 | 4.657600 | 0.000140 | 0.000017 | 1.000000e-12 | 0.05 | 3.133634 | 3.264093 | 1.026415 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | True | -2.026826 | ENSG00000104825 | hg38 | lib_1105 |
5 rows × 23 columns
We will use external gene annotations data base to extend our data
gene_annotations = sc.queries.biomart_annotations(
org="hsapiens",
attrs=["ensembl_gene_id", "gene_biotype"],
)
gene_annotations.index = gene_annotations["ensembl_gene_id"]
gene_annotation_dict = gene_annotations["gene_biotype"].to_dict()
# gene_annotations.head()
de_change["Biotype"] = [gene_annotation_dict.pop(i, "Unannotated") for i in de_change["gene_id"]]
de_change["Biotype"].value_counts()
Biotype
protein_coding 2763
lncRNA 89
transcribed_unprocessed_pseudogene 4
Unannotated 3
Mt_rRNA 2
misc_RNA 1
transcribed_unitary_pseudogene 1
Name: count, dtype: int64
display volcano plot Harringtonine vs controls volcano plot for a specific cell line and cycle
import plotnine as p9
(
p9.ggplot(de_change, p9.aes("lfc_mean", "-log10_pscore", color="Biotype"))
+ p9.geom_point(
de_change.query("Biotype == 'protein_coding'"), alpha=0.5
) # Plot other genes with transparence
+ p9.xlim(-5, 5) # Set x limits
+ p9.ylim(0, 2.5) # Set y limits
+ p9.geom_point(de_change.query("Biotype != 'protein_coding'"))
+ p9.labs(x="LFC mean", y="Significance score (higher is more significant)")
)

Display generated counts from scVI model, for top 4 genes
upregulated_genes = de_change.loc[de_change["lfc_median"] > 0, ["gene_id"]].head(4)
upregulated_genes
| gene_id | |
|---|---|
| gene_name | |
| CXCL3 | ENSG00000163734 |
| KLF10 | ENSG00000155090 |
| PIM3 | ENSG00000198355 |
| NFKBIB | ENSG00000104825 |
upregulated_genes.index
Index(['CXCL3', 'KLF10', 'PIM3', 'NFKBIB'], dtype='object', name='gene_name')
# get the generated expression from the minified model (will take very long time)
# tahoe.adata[:, upregulated_genes.index] = tahoe.get_normalized_expression(
# gene_list=list(upregulated_genes.index), n_samples=10
# )
Future Work#
Perform DE between each cell line and/or drug vs all other cell lines and/or drigs and make a dotplot of the result. In order to do this we will have to use a subset of data (both cells and genes) to save time.
Run advance models on this data such as SCANVI, MrVI using the AnnCollector dataloader.