ResolVI to address noise and biases in spatial transcriptomics#
In this tutorial, we go through the steps of training resolVI for correction in cellular-resolved spatial transcriptomics. This addresses erroneous co-expression pattern after cellular segmentation as well as unspecific background in these technologies. We highly recommend to optimize cell segmentation before running resolVI as a better cell segmentation will also yield better downstream results and both steps are complementary.
Plan for this tutorial:
Loading the data
Training an unsupervised and (semi-)supervised resolVI model
Visualizing the latent space
Transfer mapping.
Generating corrected counts and perform DE analysis
# Install from GitHub for now
!pip install --quiet scvi-colab
!pip install --quiet decoupler
!pip install --quiet adjustText
from scvi_colab import install
install()
import os
import tempfile
import decoupler as dc
import matplotlib.pyplot as plt
import numpy as np
import scanpy as sc
import scvi
sc.set_figure_params(figsize=(4, 4))
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={'facecolor' : "w"}
%config InlineBackend.figure_format='retina'
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.2
Data Loading#
For the purposes of this notebook, we will be loading a Xenium dataset of a mouse brain. We will use the left hemisphere for model training and the right hemisphere for transfer mapping. The data was originally labeled using Leiden clustering of an optimized segmentation using the ProSeg algorithm, which is a scalable algorithm for transcriptome-informed segmentation.
adata_path = os.path.join(save_dir.name, "tutorial_xenium_brain.h5ad")
adata = sc.read(
adata_path, backup_url="https://exampledata.scverse.org/scvi-tools/tutorial_xenium_brain.h5ad"
)
adata
AnnData object with n_obs × n_vars = 130604 × 248
obs: 'x_centroid', 'y_centroid', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'fov', 'predicted_celltype'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'log1p'
obsm: 'X_resolvi_transferred', 'X_spatial', 'X_umap_transferred'
layers: 'counts'
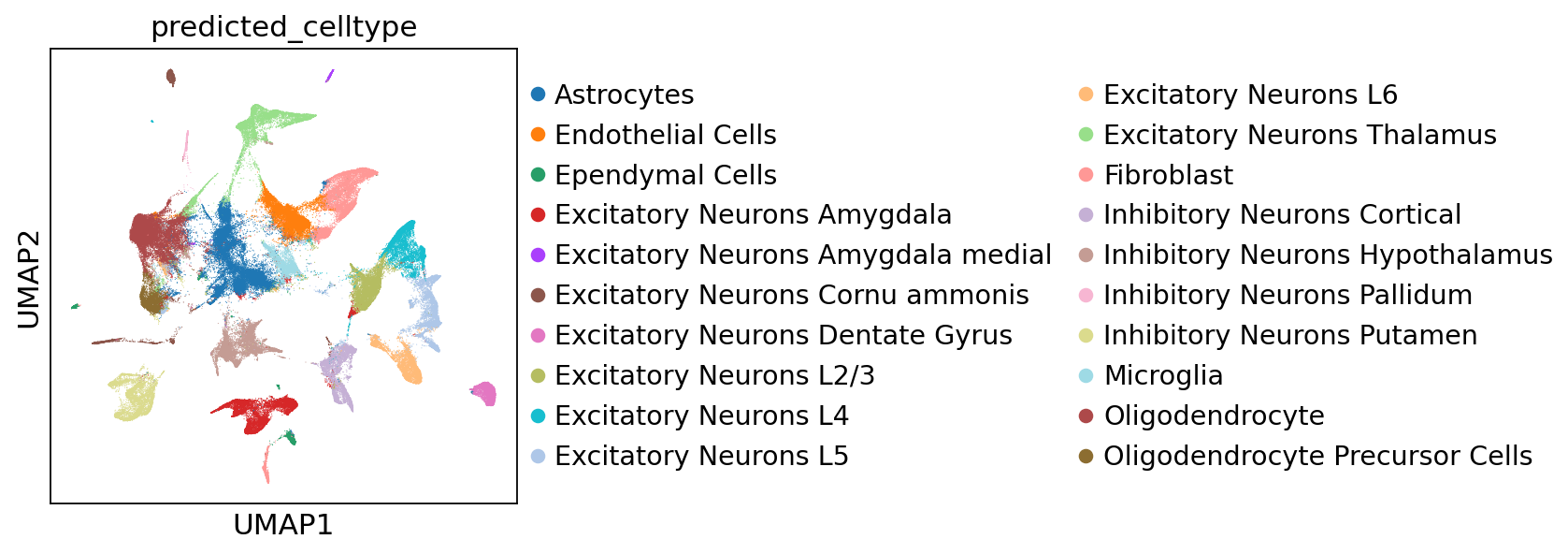
We computed a UMAP after transfering labels using the ProSeg segmented dataset and the UMAP serves as ground truth.
adata.obsm["X_umap"] = adata.obsm["X_umap_transferred"]
sc.pl.umap(adata, color="predicted_celltype")
sc.pl.spatial(adata, color="predicted_celltype", spot_size=30)

adata.obs["predicted_celltype"].value_counts()
predicted_celltype
Astrocytes 17315
Oligodendrocyte 14567
Fibroblast 10070
Endothelial Cells 9775
Inhibitory Neurons Hypothalamus 9284
Excitatory Neurons Amygdala 8640
Excitatory Neurons Thalamus 7947
Excitatory Neurons L5 7004
Excitatory Neurons L2/3 6393
Inhibitory Neurons Putamen 5753
Inhibitory Neurons Cortical 5711
Excitatory Neurons L4 5500
Excitatory Neurons L6 5052
Microglia 4141
Oligodendrocyte Precursor Cells 3872
Excitatory Neurons Dentate Gyrus 3283
Excitatory Neurons Cornu ammonis 2787
Ependymal Cells 2202
Inhibitory Neurons Pallidum 658
Excitatory Neurons Amygdala medial 650
Name: count, dtype: int64
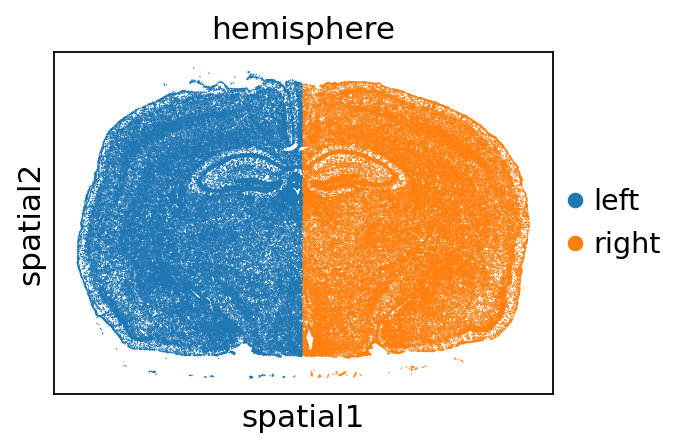
We use this dataset to set up a semisupervised and an unsupervised model in resolVI and train these. We split the dataset into right and left hemisphere to demonstrate query mapping.
adata.obs["hemisphere"] = ["right" if x > 5000 else "left" for x in adata.obsm["X_spatial"][:, 0]]
sc.pl.spatial(adata, color="hemisphere", spot_size=30)
ref_adata = adata[adata.obs["hemisphere"] == "left"].copy()
query_adata = adata[adata.obs["hemisphere"] == "right"].copy()
Train resolVI model#
As in the scANVI notebook, we need to register the AnnData object for use in resolVI. Namely, we can ignore the batch parameter because those cells don’t have much batch effect to begin with as they are derived from a single slice. However, we will give the celltype labels for resolVI to use. Setting up AnnData computes spatial neighbors within each batch. This step might take minutes for very large datasets. It is important that different slices are used as batch covariate.
scvi.external.RESOLVI.setup_anndata(ref_adata, labels_key="predicted_celltype", layer="counts")
INFO Generating sequential column names
INFO Generating sequential column names
supervised_resolvi = scvi.external.RESOLVI(ref_adata, semisupervised=True)
We use here only 20 epochs to speed up running the tutorial. We would recommend using 100 epochs here.
supervised_resolvi.train(max_epochs=50)
Now we can predict the cell type labels using the trained model, and get the latent space. ResolVI uses pyro. Pyro stores parameters in a pyro_param_store. It is necessary that only a single model is used at a time, e.g. after query training only the query model is valid and the reference model gets overwritten.
ref_adata
AnnData object with n_obs × n_vars = 66457 × 248
obs: 'x_centroid', 'y_centroid', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'fov', 'predicted_celltype', 'hemisphere', '_indices', '_scvi_batch', '_scvi_ind_x', '_scvi_labels'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'log1p', 'predicted_celltype_colors', 'hemisphere_colors', '_scvi_uuid', '_scvi_manager_uuid'
obsm: 'X_resolvi_transferred', 'X_spatial', 'X_umap_transferred', 'X_umap', 'index_neighbor', 'distance_neighbor'
layers: 'counts'
ref_adata.obsm["resolvi_celltypes"] = supervised_resolvi.predict(
ref_adata, num_samples=3, soft=True
)
ref_adata.obs["resolvi_predicted"] = ref_adata.obsm["resolvi_celltypes"].idxmax(axis=1)
ref_adata.obsm["X_resolVI"] = supervised_resolvi.get_latent_representation(ref_adata)
Again, we may visualize the latent space as well as the inferred labels
sc.pp.neighbors(ref_adata, use_rep="X_resolVI")
sc.tl.umap(ref_adata)
ref_adata
AnnData object with n_obs × n_vars = 66457 × 248
obs: 'x_centroid', 'y_centroid', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'fov', 'predicted_celltype', 'hemisphere', '_indices', '_scvi_batch', '_scvi_ind_x', '_scvi_labels', 'resolvi_predicted'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'log1p', 'predicted_celltype_colors', 'hemisphere_colors', '_scvi_uuid', '_scvi_manager_uuid', 'neighbors', 'umap'
obsm: 'X_resolvi_transferred', 'X_spatial', 'X_umap_transferred', 'X_umap', 'index_neighbor', 'distance_neighbor', 'resolvi_celltypes', 'X_resolVI'
layers: 'counts'
obsp: 'distances', 'connectivities'
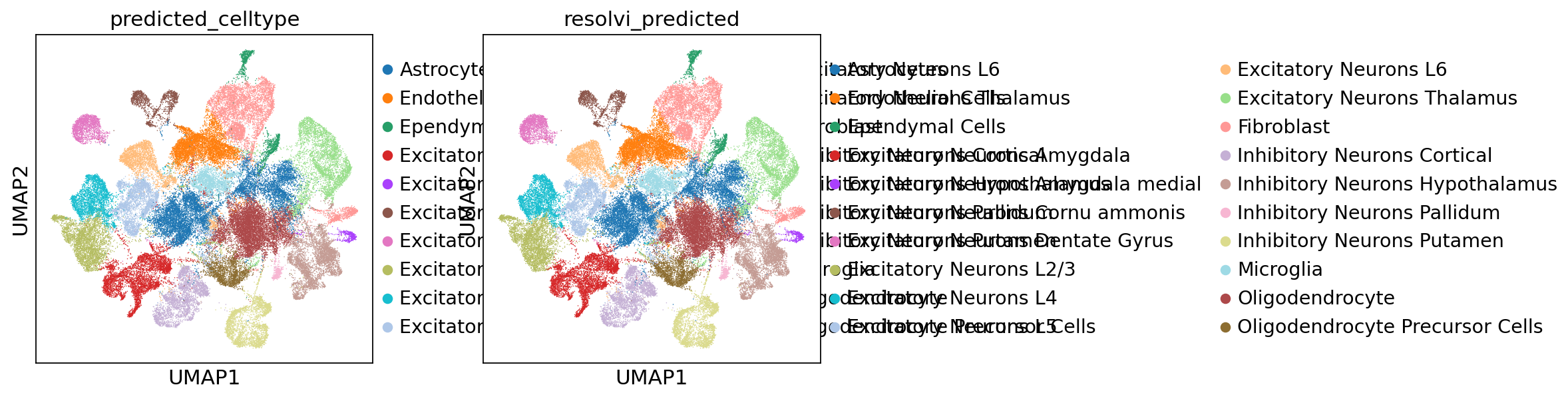
sc.pl.umap(ref_adata, color=["predicted_celltype", "resolvi_predicted"])
sc.pl.spatial(ref_adata, color=["predicted_celltype", "resolvi_predicted"], spot_size=30)

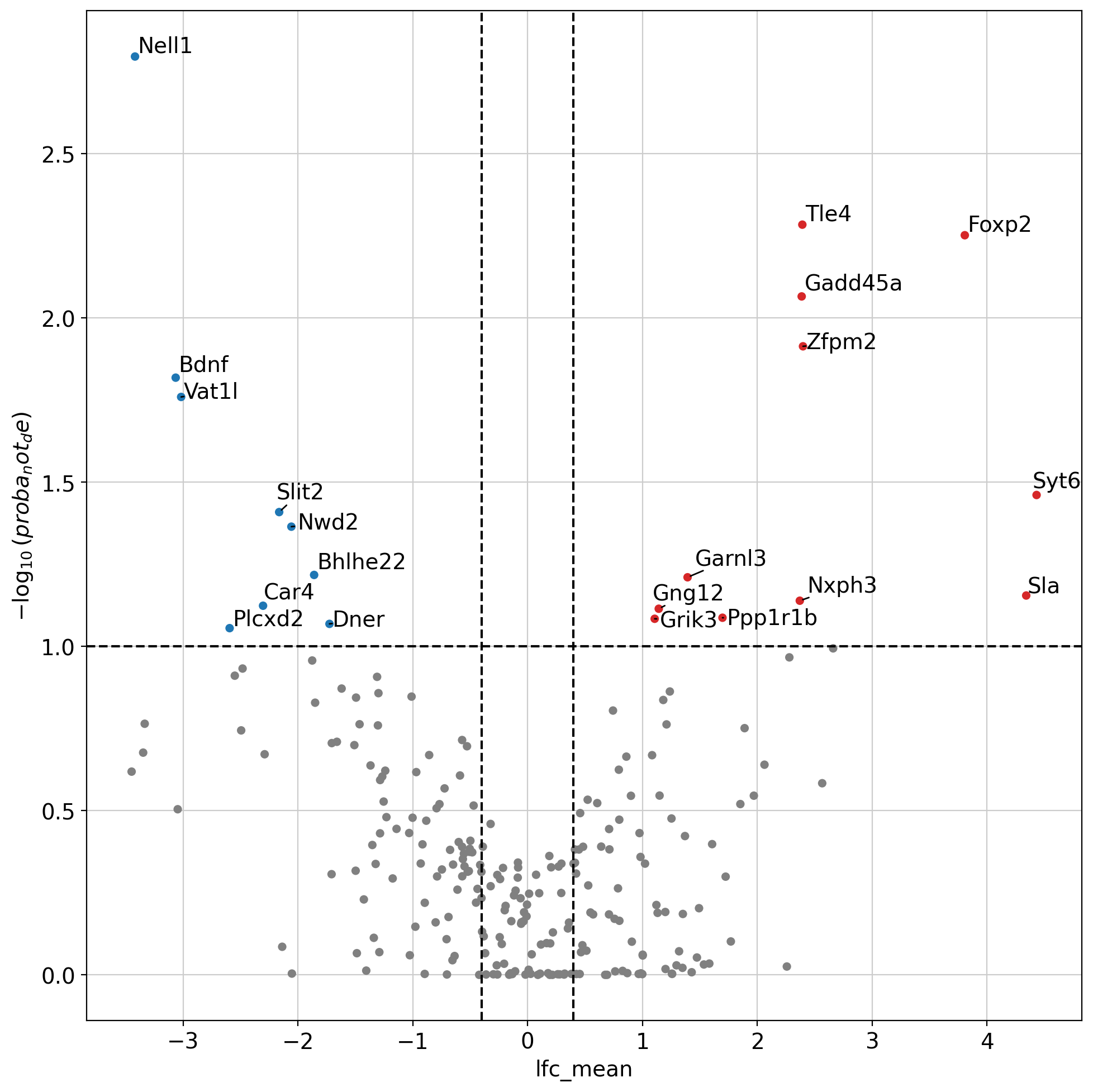
We can use the trained model to perform differential expression of two groups of cells. We compute here genes differentially expressed between neurons in two distinct layers. This uses a similar test to the scVI DE test. Please keep in mind this test doesn’t test for differences in the mean between two groups but tests for differences between random pairs of single cell.
de_result_importance = supervised_resolvi.differential_expression(
ref_adata,
groupby="resolvi_predicted",
group1="Excitatory Neurons L6",
group2="Excitatory Neurons L5",
weights="importance",
pseudocounts=1e-2,
delta=0.05,
filter_outlier_cells=True,
mode="change",
test_mode="three",
)
de_result_importance.head(5)
| proba_de | proba_not_de | bayes_factor | scale1 | scale2 | pseudocounts | delta | lfc_mean | lfc_median | lfc_std | ... | raw_mean1 | raw_mean2 | non_zeros_proportion1 | non_zeros_proportion2 | raw_normalized_mean1 | raw_normalized_mean2 | is_de_fdr_0.05 | comparison | group1 | group2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nell1 | 0.9984 | 0.0016 | 6.436144 | 0.000669 | 0.007273 | 0.01 | 0.05 | -3.419098 | -3.474191 | 0.931534 | ... | 0.322696 | 2.675334 | 0.239729 | 0.649358 | 10.863630 | 69.241287 | True | Excitatory Neurons L6 vs Excitatory Neurons L5 | Excitatory Neurons L6 | Excitatory Neurons L5 |
| Tle4 | 0.9948 | 0.0052 | 5.253881 | 0.027108 | 0.005897 | 0.01 | 0.05 | 2.392308 | 2.467812 | 0.838471 | ... | 7.932221 | 2.020828 | 0.980056 | 0.719544 | 259.170776 | 57.704887 | True | Excitatory Neurons L6 vs Excitatory Neurons L5 | Excitatory Neurons L6 | Excitatory Neurons L5 |
| Foxp2 | 0.9944 | 0.0056 | 5.179371 | 0.012461 | 0.001021 | 0.01 | 0.05 | 3.807136 | 3.822906 | 1.385957 | ... | 3.219745 | 0.400570 | 0.814918 | 0.263909 | 103.027725 | 11.164698 | True | Excitatory Neurons L6 vs Excitatory Neurons L5 | Excitatory Neurons L6 | Excitatory Neurons L5 |
| Gadd45a | 0.9914 | 0.0086 | 4.747355 | 0.016747 | 0.003301 | 0.01 | 0.05 | 2.386628 | 2.424568 | 0.869711 | ... | 5.288797 | 1.195151 | 0.901476 | 0.521255 | 170.523941 | 32.507549 | True | Excitatory Neurons L6 vs Excitatory Neurons L5 | Excitatory Neurons L6 | Excitatory Neurons L5 |
| Zfpm2 | 0.9878 | 0.0122 | 4.394043 | 0.005375 | 0.001195 | 0.01 | 0.05 | 2.398096 | 2.344832 | 1.027264 | ... | 1.726348 | 0.380885 | 0.708416 | 0.261626 | 56.197052 | 10.260593 | True | Excitatory Neurons L6 vs Excitatory Neurons L5 | Excitatory Neurons L6 | Excitatory Neurons L5 |
5 rows × 22 columns
dc.pl.volcano(
de_result_importance,
x="lfc_mean",
y="proba_not_de",
thr_sign=0.1,
thr_stat=0.4,
top=30,
figsize=(10, 10),
)
plt.show()
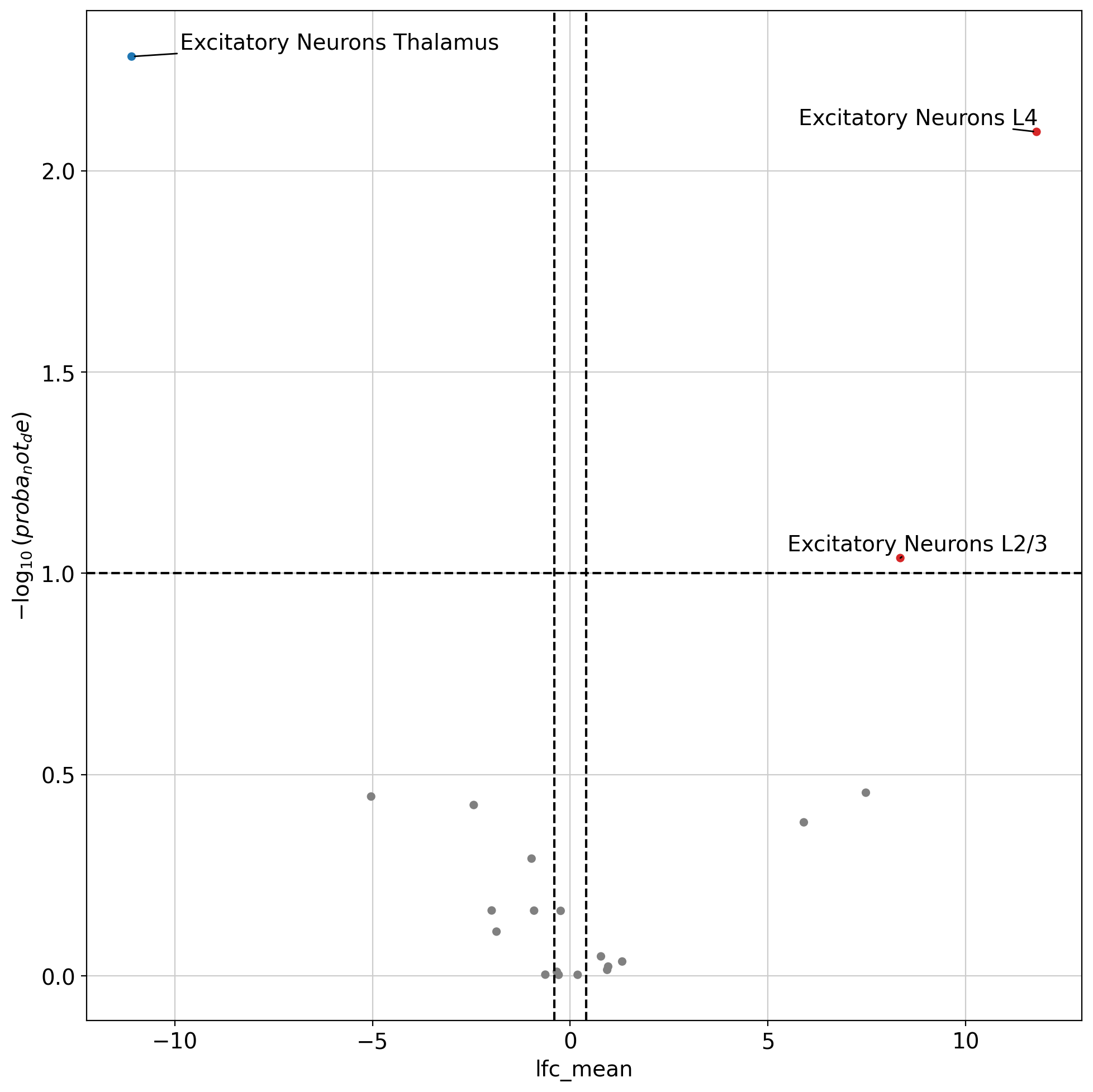
We can use the trained model to perform differential abundance of cell states in the neighborhood of two groups of cells. We find here that excitatory neurons in thalamus and cortex preferentially colocalize with themselves as well as adjacent layer neurons. This uses a similar test to the scVI DE test.
da = supervised_resolvi.differential_niche_abundance(
groupby="resolvi_predicted",
group1="Excitatory Neurons L4",
group2="Excitatory Neurons Thalamus",
neighbor_key="index_neighbor",
test_mode="three",
delta=0.05,
pseudocounts=3e-2,
)
da.head(5)
| proba_de | proba_not_de | bayes_factor | scale1 | scale2 | pseudocounts | delta | lfc_mean | lfc_median | lfc_std | lfc_min | lfc_max | is_de_fdr_0.05 | comparison | group1 | group2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Excitatory Neurons Thalamus | 0.9948 | 0.0052 | 5.253881 | 0.002442 | 0.537802 | 0.03 | 0.05 | -11.090903 | -11.991620 | 2.943154 | -14.944863 | 9.273638 | True | Excitatory Neurons L4 vs Excitatory Neurons Th... | Excitatory Neurons L4 | Excitatory Neurons Thalamus |
| Excitatory Neurons L4 | 0.9920 | 0.0080 | 4.820280 | 0.484913 | 0.000780 | 0.03 | 0.05 | 11.791515 | 12.180008 | 1.938235 | -1.341406 | 14.845583 | True | Excitatory Neurons L4 vs Excitatory Neurons Th... | Excitatory Neurons L4 | Excitatory Neurons Thalamus |
| Excitatory Neurons L2/3 | 0.9084 | 0.0916 | 2.294253 | 0.077645 | 0.000099 | 0.03 | 0.05 | 8.346159 | 8.289958 | 3.097958 | -5.414918 | 14.874312 | True | Excitatory Neurons L4 vs Excitatory Neurons Th... | Excitatory Neurons L4 | Excitatory Neurons Thalamus |
| Inhibitory Neurons Cortical | 0.6494 | 0.3506 | 0.616403 | 0.088365 | 0.000517 | 0.03 | 0.05 | 7.474957 | 10.954712 | 5.380948 | -13.136246 | 14.186956 | False | Excitatory Neurons L4 vs Excitatory Neurons Th... | Excitatory Neurons L4 | Excitatory Neurons Thalamus |
| Oligodendrocyte | 0.6416 | 0.3584 | 0.582315 | 0.021661 | 0.080403 | 0.03 | 0.05 | -5.034796 | -5.462290 | 6.335117 | -14.455128 | 12.528509 | False | Excitatory Neurons L4 vs Excitatory Neurons Th... | Excitatory Neurons L4 | Excitatory Neurons Thalamus |
dc.pl.volcano(
da,
x="lfc_mean",
y="proba_not_de",
thr_sign=0.1,
thr_stat=0.4,
top=30,
figsize=(10, 10),
)
plt.show()
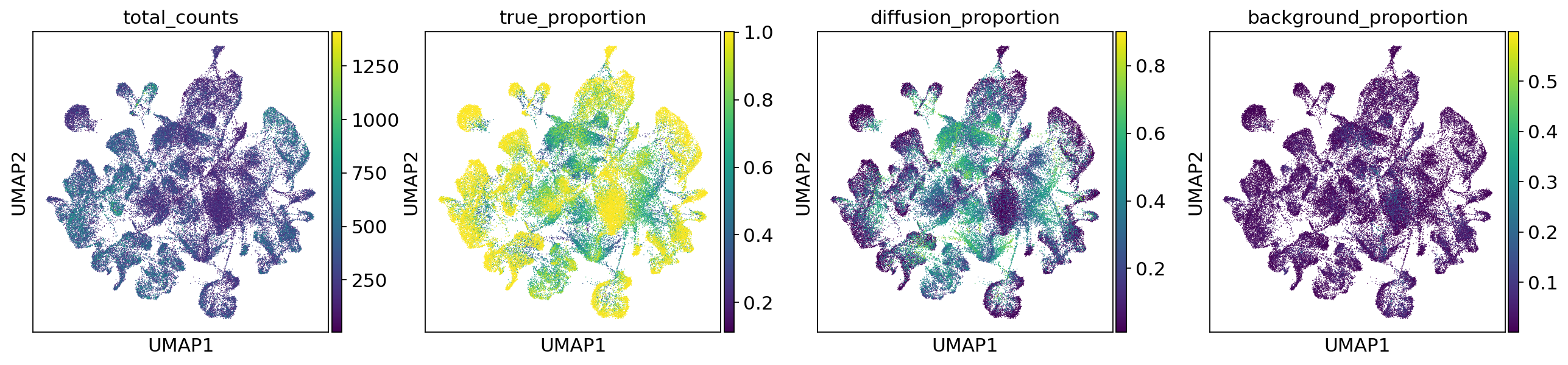
We can also generate counts that are corrected for counts from neighboring cells wrongly assigned due to missegmentation as well as unspecific background. We use custom parameters for num_samples and and summary_fun to accelerate computation. “px_rate” of model_corrected generates corrected count data. “mixture_proportions” of model_residuals generates the amount of diffusion and background for each cell. Batch steps defines how many batches are accumulated before computing summary statistics. To increase the amount of correction, use lower quantiles instead of median.
import pandas as pd
samples_corr = supervised_resolvi.sample_posterior(
model=supervised_resolvi.module.model_corrected,
return_sites=["px_rate"],
summary_fun={"post_sample_q50": np.median},
num_samples=3,
summary_frequency=30,
)
samples_corr = pd.DataFrame(samples_corr).T
samples = supervised_resolvi.sample_posterior(
model=supervised_resolvi.module.model_residuals,
return_sites=["mixture_proportions"],
summary_fun={"post_sample_means": np.mean},
num_samples=3,
summary_frequency=100,
)
samples = pd.DataFrame(samples).T
ref_adata.obs[["true_proportion", "diffusion_proportion", "background_proportion"]] = samples.loc[
"post_sample_means", "mixture_proportions"
]
sc.pl.umap(
ref_adata,
color=["total_counts", "true_proportion", "diffusion_proportion", "background_proportion"],
)
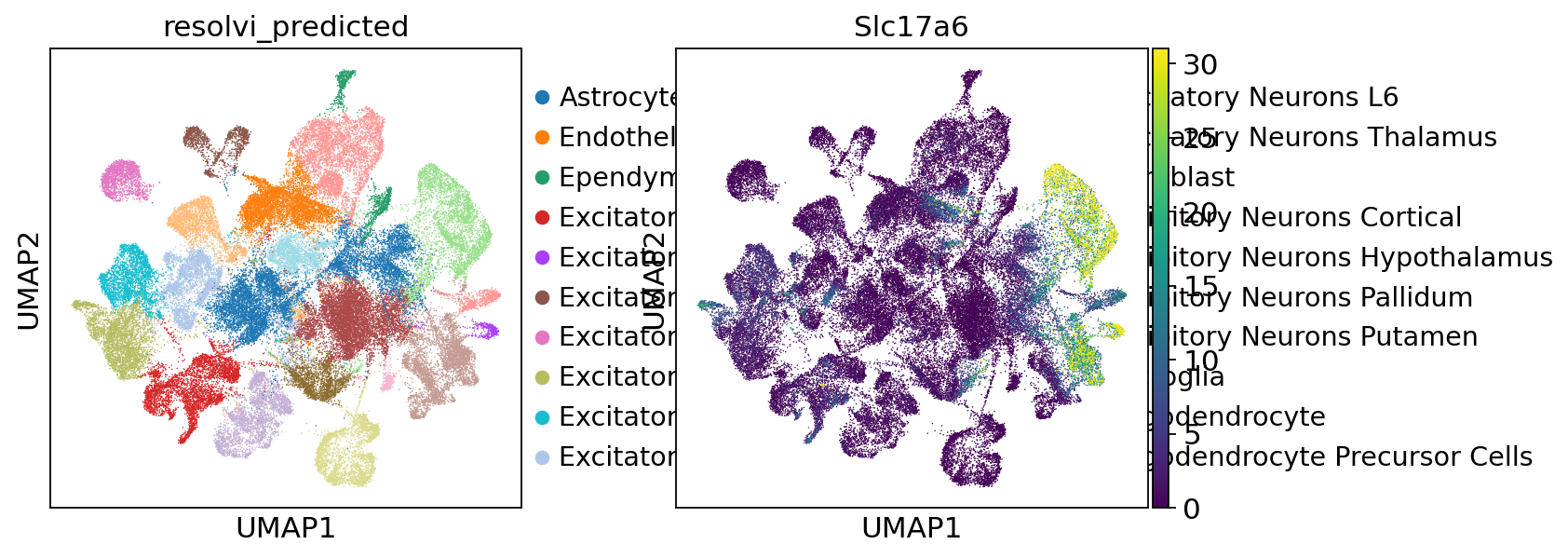
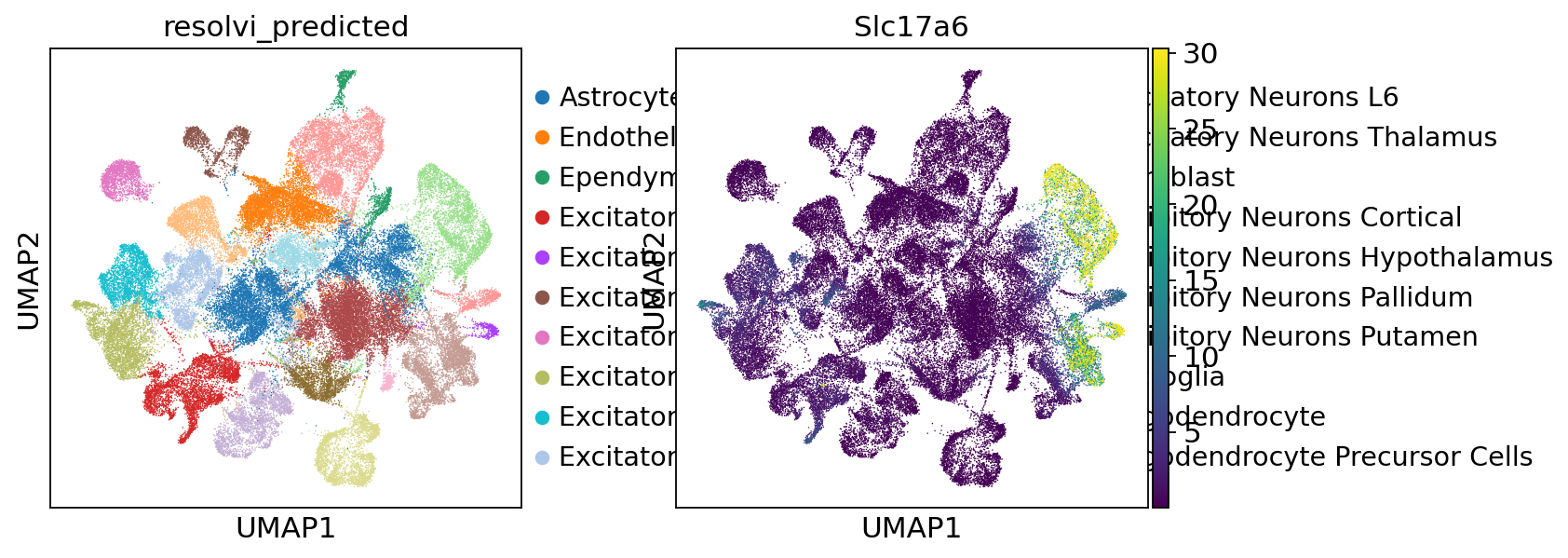
ref_adata.layers["generated_expression"] = samples_corr.loc["post_sample_q50", "px_rate"]
sc.pl.umap(ref_adata, color=["resolvi_predicted", "Slc17a6"], layer="counts", vmax="p98")
sc.pl.umap(
ref_adata, color=["resolvi_predicted", "Slc17a6"], layer="generated_expression", vmax="p98"
)
Query transfer#
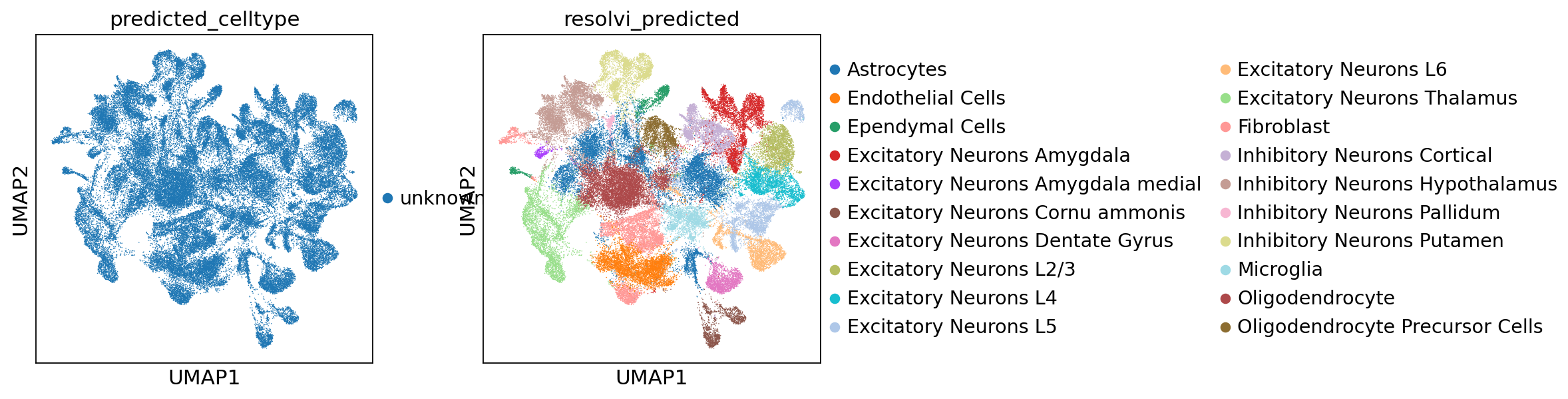
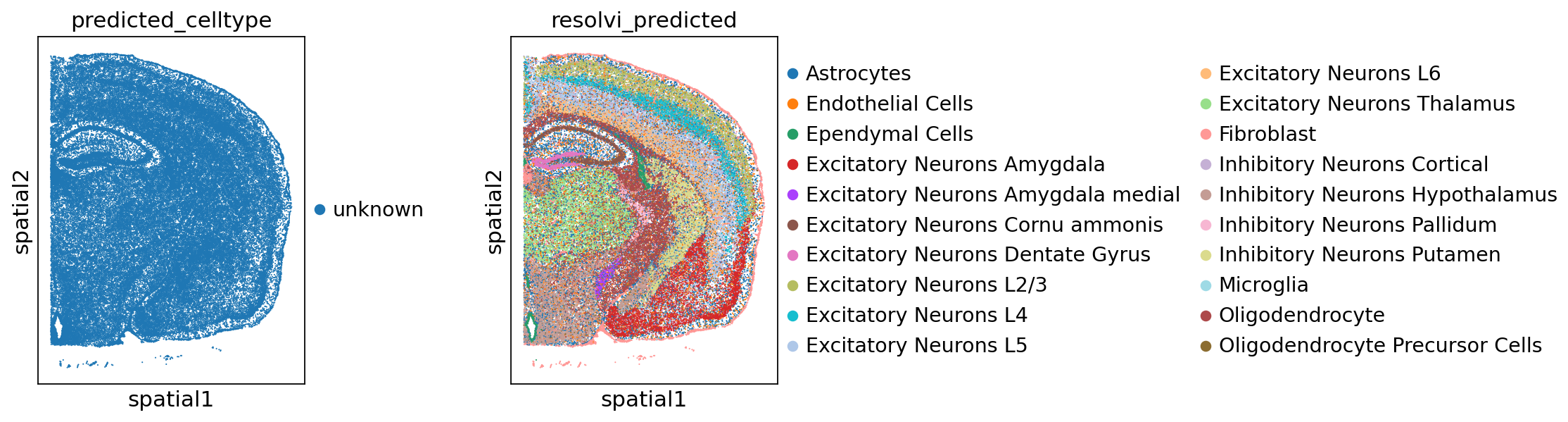
We can train the reference model on query data to reannotate this data. We rely on the observation names for non-amortized parameters in resolVI. It is important to make sure cells have unique names between query and reference dataset. We set the cell-type to unknown, so that resolVI needs to predict the celltype leveraging the reference model.
query_adata.obs["predicted_celltype"] = "unknown"
query_adata.obs_names = [f"query_{i}" for i in query_adata.obs_names]
supervised_resolvi.prepare_query_anndata(query_adata, reference_model=supervised_resolvi)
query_resolvi = supervised_resolvi.load_query_data(query_adata, reference_model=supervised_resolvi)
INFO Found 100.0% reference vars in query data.
query_resolvi.train(max_epochs=20)
Running scArches. Set lr to 0 and blocking variables.
query_adata.obs["resolvi_predicted"] = query_resolvi.predict(
query_adata, num_samples=3, soft=False
)
query_adata.obsm["X_resolVI"] = query_resolvi.get_latent_representation(query_adata)
Again, we may visualize the latent space as well as the inferred labels
sc.pp.neighbors(query_adata, use_rep="X_resolVI")
sc.tl.umap(query_adata)
sc.pl.umap(query_adata, color=["predicted_celltype", "resolvi_predicted"])
sc.pl.spatial(query_adata, color=["predicted_celltype", "resolvi_predicted"], spot_size=30)
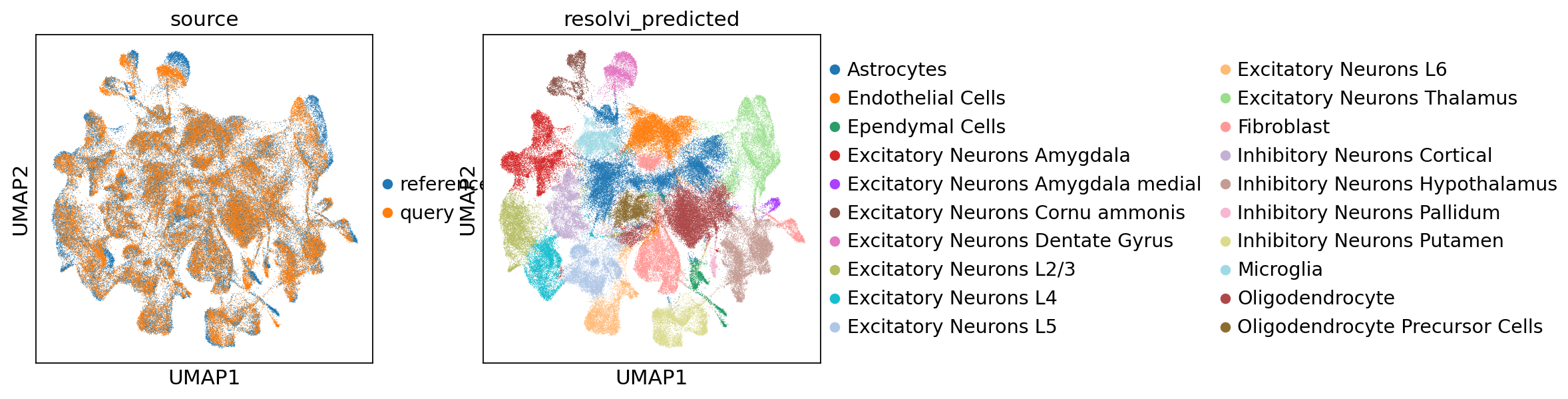
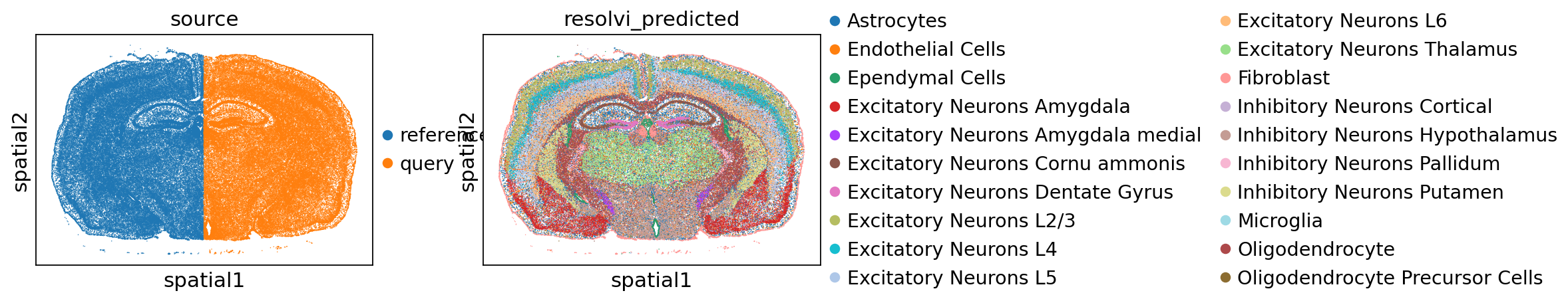
We can now concatenate the datasets again and find good integration and accurate cell-type information.
full_adata = ref_adata.concatenate(
query_adata, batch_key="source", batch_categories=["reference", "query"]
)
sc.pp.neighbors(full_adata, use_rep="X_resolVI")
sc.tl.umap(full_adata)
sc.pl.umap(full_adata, color=["source", "resolvi_predicted"])
sc.pl.spatial(full_adata, color=["source", "resolvi_predicted"], spot_size=30)