Using SHAP values and IntegratedGradients for cell type classification interpretability#
Previously we saw semi-supervised models, like SCANVI being used for tasks like cell type classification, enabling researchers to uncover complex biological patterns. However, as these models become more sophisticated, it is essential to understand not just the predictions they make, but why they make them. This is where interpretability methods like SHAP (SHapley Additive exPlanations) and CAPTUM IntegratedGradients come into play. By providing insights into the influence of individual features on model predictions, these methods help us trust and validate our models in critical biological contexts.
In this tutorial, we’ll explore the significance of interpretability techniques in supervised cell classification using ScanVI, which are now avialble as part of SCVI-Tools.
Note
Running the following cell will install tutorial dependencies on Google Colab only. It will have no effect on environments other than Google Colab.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import seaborn as sns
import torch
torch.set_float32_matmul_precision("high")
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.4.2
Load data and train scanvi#
In this tutorial we will be using the dataset of peripheral blood mononuclear cells from 10x Genomics, PBMC dataset
adata = scvi.data.pbmc_dataset()
adata.layers["counts"] = adata.X.copy()
adata.obs["batch"] = adata.obs["batch"].astype("category")
adata
INFO File data/gene_info_pbmc.csv already downloaded
INFO File data/pbmc_metadata.pickle already downloaded
INFO File data/pbmc8k/filtered_gene_bc_matrices.tar.gz already downloaded
INFO Extracting tar file
INFO Removing extracted data at data/pbmc8k/filtered_gene_bc_matrices
INFO File data/pbmc4k/filtered_gene_bc_matrices.tar.gz already downloaded
INFO Extracting tar file
INFO Removing extracted data at data/pbmc4k/filtered_gene_bc_matrices
AnnData object with n_obs × n_vars = 11990 × 3346
obs: 'n_counts', 'batch', 'labels', 'str_labels'
var: 'gene_symbols', 'n_counts-0', 'n_counts-1', 'n_counts'
uns: 'cell_types'
obsm: 'design', 'raw_qc', 'normalized_qc', 'qc_pc'
layers: 'counts'
adata.var_names = adata.var["gene_symbols"]
adata.obs.str_labels.value_counts() # list of classes and their observations
str_labels
CD4 T cells 4996
CD14+ Monocytes 2227
B cells 1621
CD8 T cells 1448
Other 463
NK cells 457
FCGR3A+ Monocytes 351
Dendritic Cells 339
Megakaryocytes 88
Name: count, dtype: int64
print("# cells, # genes before filtering:", adata.shape)
sc.pp.filter_genes(adata, min_counts=3)
sc.pp.filter_cells(adata, min_counts=3)
# cells, # genes before filtering: (11990, 3346)
# We select a small number of genes here, so our later interpretability analysis will be fast
sc.pp.highly_variable_genes(
adata,
n_top_genes=200,
subset=True,
layer="counts",
flavor="seurat_v3",
batch_key="batch",
)
print("# cells, # genes after filtering:", adata.shape)
# cells, # genes after filtering: (11990, 200)
scvi.model.SCANVI.setup_anndata(
adata,
layer="counts",
batch_key="batch",
labels_key="str_labels",
unlabeled_category="unknown",
)
model = scvi.model.SCANVI(adata)
model
ScanVI Model with the following params: unlabeled_category: unknown, n_hidden: 128, n_latent: 10, n_layers: 1, dropout_rate: 0.1, dispersion: gene, gene_likelihood: zinb Training status: Not Trained Model's adata is minified?: False
model.train(
max_epochs=100,
early_stopping=True,
check_val_every_n_epoch=1,
train_size=0.8,
validation_size=0.2,
# accelerator="gpu",
# devices=-1,
# strategy="ddp_notebook_find_unused_parameters_true",
)
INFO Training for 100 epochs.
Inspect scanvi training and test performance#
adata.obsm["X_scANVI"] = model.get_latent_representation()
# use scVI latent space for UMAP generation
sc.pp.neighbors(adata, use_rep="X_scANVI", n_neighbors=30)
sc.tl.umap(adata, min_dist=0.3)
sc.pl.umap(adata, color=["str_labels", "batch"], ncols=2, wspace=0.4)

Next we will apply the 2 techniques for features interpretability and compare between them
Integrated Gradients#
Integrated Gradients is a robust interpretability technique that attributes the output of a model to its input features by calculating the cumulative sum of gradients along a path from a baseline (typically zero or a neutral input) to the actual input. This approach provides a way to measure how each feature contributes to the model’s output in a smooth and consistent manner.
It is availble for any semi supervised model in SCVI-Tools by passing the ig_interpretability=True flag to the predict function.
predictions, attributions = model.predict(ig_interpretability=True)
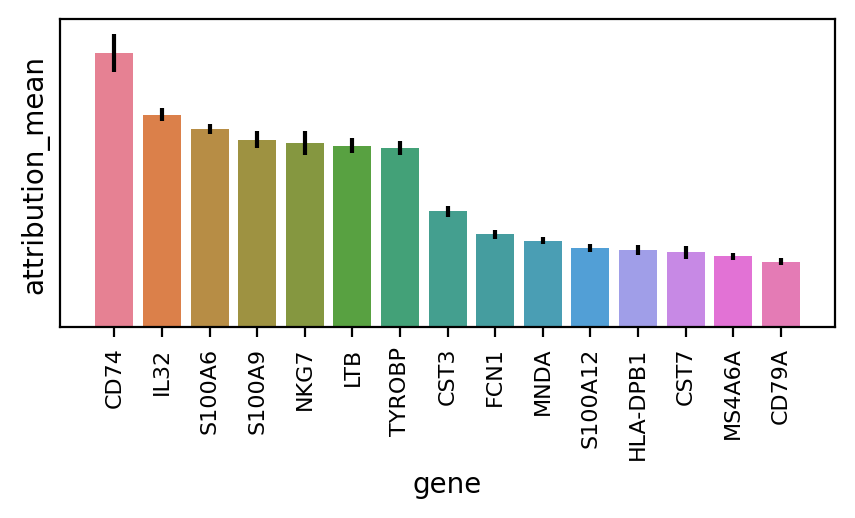
The method works relatievely fast and we can then plot the gene table with their importnace mean and variance, overall for all cell - types
n_plot = 15
attributions.head(n_plot)
| gene | gene_idx | attribution_mean | attribution_std | cells | |
|---|---|---|---|---|---|
| 0 | CD74 | 70 | 1.046176 | 4.104647 | 11990 |
| 1 | IL32 | 148 | 0.810820 | 1.358012 | 11990 |
| 2 | S100A6 | 17 | 0.756088 | 1.103666 | 11990 |
| 3 | S100A9 | 15 | 0.716369 | 1.809122 | 11990 |
| 4 | NKG7 | 186 | 0.702932 | 2.459903 | 11990 |
| 5 | LTB | 78 | 0.692312 | 1.575437 | 11990 |
| 6 | TYROBP | 179 | 0.683626 | 1.525760 | 11990 |
| 7 | CST3 | 166 | 0.441730 | 1.180127 | 11990 |
| 8 | FCN1 | 104 | 0.354472 | 0.991154 | 11990 |
| 9 | MNDA | 20 | 0.329743 | 0.744831 | 11990 |
| 10 | S100A12 | 16 | 0.302508 | 0.769542 | 11990 |
| 11 | HLA-DPB1 | 81 | 0.296287 | 1.080156 | 11990 |
| 12 | CST7 | 167 | 0.285223 | 1.292438 | 11990 |
| 13 | MS4A6A | 110 | 0.271008 | 0.730065 | 11990 |
| 14 | CD79A | 180 | 0.250256 | 0.732563 | 11990 |
df = attributions.head(n_plot)
ci = 1.96 * df["attribution_std"] / np.sqrt(df["cells"])
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 2), dpi=200)
sns.barplot(ax=ax, data=df, x="gene", y="attribution_mean", hue="gene", dodge=False)
ax.set_yticks([])
plt.tick_params(axis="x", which="major", labelsize=8, labelrotation=90)
ax.errorbar(
df["gene"].values,
df["attribution_mean"].values,
yerr=ci,
ecolor="black",
fmt="none",
)
if ax.get_legend() is not None:
ax.get_legend().remove()
We can repeat for specific class (‘Dendritic Cells’):
predictions_class, attributions_class = model.predict(
indices=np.where(adata.obs.str_labels == "Dendritic Cells")[0].tolist(),
ig_interpretability=True,
)
attributions_class.head(n_plot)
| gene | gene_idx | attribution_mean | attribution_std | cells | |
|---|---|---|---|---|---|
| 0 | CD74 | 70 | 5.473364 | 0.948142 | 339 |
| 1 | FCER1A | 21 | 3.630613 | 1.943166 | 339 |
| 2 | HLA-DPB1 | 81 | 3.318439 | 0.631406 | 339 |
| 3 | VIM | 117 | 1.793772 | 0.506637 | 339 |
| 4 | ANXA1 | 97 | 1.650854 | 0.695445 | 339 |
| 5 | HLA-DMB | 79 | 1.374406 | 0.584015 | 339 |
| 6 | CLEC10A | 157 | 1.342435 | 1.255666 | 339 |
| 7 | HLA-DMA | 80 | 1.158052 | 0.371506 | 339 |
| 8 | COTL1 | 154 | 1.053837 | 0.387444 | 339 |
| 9 | TYROBP | 179 | 0.987148 | 0.421023 | 339 |
| 10 | S100A10 | 13 | 0.952728 | 0.462643 | 339 |
| 11 | ALDH2 | 130 | 0.767798 | 0.441154 | 339 |
| 12 | PHACTR1 | 75 | 0.702436 | 0.534552 | 339 |
| 13 | CAPG | 31 | 0.656638 | 0.390209 | 339 |
| 14 | LGALS3 | 141 | 0.514238 | 0.382886 | 339 |
df_class = attributions_class.head(n_plot)
ci = 1.96 * df_class["attribution_std"] / np.sqrt(df_class["cells"])
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 2), dpi=200)
sns.barplot(ax=ax, data=df_class, x="gene", y="attribution_mean", hue="gene", dodge=False)
ax.set_yticks([])
plt.tick_params(axis="x", which="major", labelsize=8, labelrotation=90)
ax.errorbar(
df_class["gene"].values,
df_class["attribution_mean"].values,
yerr=ci,
ecolor="black",
fmt="none",
)
if ax.get_legend() is not None:
ax.get_legend().remove()

As expected, for a specific class, we can see different important genes, altough S100A4 is still the top contributer
More generally we would like to see a more general view of top genes to contribute to our celltype groups classification.
classes = adata.obs.str_labels.cat.categories
features = adata.var_names
attributions_class_pos_agg = pd.DataFrame()
n_cols = 3
top_n = 5
nrows = round(classes.size / n_cols)
fig, ax = plt.subplots(nrows, n_cols, sharex=False, figsize=(20, 20))
for idx, ct in enumerate(classes):
_, attributions_class = model.predict(
indices=np.where(adata.obs.str_labels == ct)[0].tolist(),
ig_interpretability=True,
)
positive = attributions_class.head(top_n)
positive["contribution"] = "positive"
negative = attributions_class.tail(top_n)
negative["contribution"] = "negative"
avg = pd.concat([positive, negative])
title = f"IG importance for: {ct}"
# also keep the positive contributions
attributions_class_pos = attributions_class[attributions_class.attribution_mean > 0]
attributions_class_pos["class"] = ct
attributions_class_pos_agg = pd.concat([attributions_class_pos_agg, attributions_class_pos])
sns.barplot(
x="attribution_mean",
y="gene",
hue="contribution",
palette=["blue", "red"],
data=avg,
ax=ax[idx // n_cols, idx % n_cols],
)
ax[idx // n_cols, idx % n_cols].set_title(title)
ax[idx // n_cols, idx % n_cols].legend(title="IG Contribution", loc="lower right")
_ = [fig.delaxes(ax_) for ax_ in ax.flatten() if not ax_.has_data()]
fig.tight_layout()

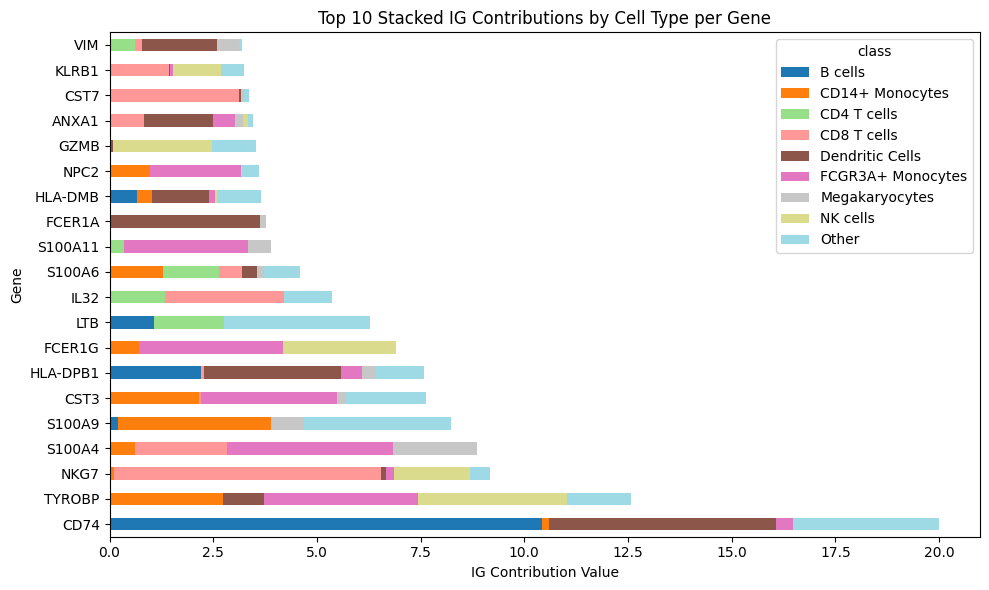
And we can also show the positive contribution of each gene being aggregated per cell type group
top_n = 20
# Pivot the data so that each group becomes a column for stacking
pivot_df = attributions_class_pos_agg.pivot_table(
index="gene", columns="class", values="attribution_mean", aggfunc="sum"
)
# Sort by the total sum of each feature (sum across all groups)
pivot_df["total"] = pivot_df.sum(axis=1) # Calculate the total sum for each feature
pivot_df = pivot_df.sort_values(by="total", ascending=False) # Sort by the total value
pivot_df = pivot_df.head(top_n) # Select the top 10 features
# Plotting the horizontal stacked bar plot
ax = pivot_df.drop("total", axis=1).plot(
kind="barh", stacked=True, figsize=(10, 6), colormap="tab20"
)
# Add labels and title
ax.set_xlabel("IG Contribution Value")
ax.set_ylabel("Gene")
ax.set_title("Top 10 Stacked IG Contributions by Cell Type per Gene")
# Display the plot
plt.tight_layout()
plt.show()
SHAP#
SHAP (SHapley Additive exPlanations) values are a popular interpretability technique based on cooperative game theory. The core idea is to fairly allocate the “credit” for a model’s prediction to each feature, by considering all possible combinations of features and their impact on the prediction. SHAP values are additive, meaning the sum of the SHAP values for all features equals the difference between the model’s output and the average prediction. This method works for any model type, providing a consistent way to explain individual predictions, making it highly versatile and widely applicable. Deep SHAP is an extension of the SHAP method designed specifically for deep learning models, such as the ones in SCVI-Tools. For more information see this
Calcualtion of SHAP for SC data usually takes a lot of time. In SCVI-Tools we are running an approximation of FastSHAP in order to reduce runtime, where we train a shallow surrogate model to imitate the original model prediction and than run the SHAP over the surrogate model. See this
import torch.nn as nn
from scvi.utils import FastSHAP, Surrogate
from scvi.utils.fastshap import KLDivLoss, MaskLayer1d
num_features = len(features)
num_classes = len(classes)
surr = nn.Sequential(
MaskLayer1d(value=0, append=True),
nn.Linear(2 * num_features, 128),
nn.ELU(inplace=True),
nn.Linear(128, 128),
nn.ELU(inplace=True),
nn.Linear(128, num_classes),
).to(model.device)
# Set up surrogate object
surrogate = Surrogate(surr, num_features)
# Train Surrogate
surrogate.train_original_model(
train_data=adata.X.toarray()[model.train_indices],
val_data=adata.X.toarray()[model.validation_indices],
original_model=model.shap_adata_predict,
batch_size=64,
max_epochs=10,
loss_fn=KLDivLoss(),
validation_samples=10,
validation_batch_size=10000,
verbose=True,
)
# Train FastSHAP
# Create explainer model
explainer = nn.Sequential(
nn.Linear(num_features, 128),
nn.ReLU(inplace=True),
nn.Linear(128, 128),
nn.ReLU(inplace=True),
nn.Linear(128, num_classes * num_features),
).to(model.device)
# Set up FastSHAP object
fastshap = FastSHAP(explainer, surrogate, normalization="additive", link=nn.Softmax(dim=-1))
# Train
fastshap.train(
train_data=adata.X.toarray()[model.train_indices],
val_data=adata.X.toarray()[model.validation_indices],
batch_size=32,
num_samples=32,
max_epochs=100,
validation_samples=128,
verbose=True,
)
----- Epoch = 1 -----
Val loss = 5.208660
New best epoch, loss = 5.208660
----- Epoch = 2 -----
Val loss = 1.557097
New best epoch, loss = 1.557097
----- Epoch = 3 -----
Val loss = 0.361534
New best epoch, loss = 0.361534
----- Epoch = 4 -----
Val loss = 0.076193
New best epoch, loss = 0.076193
----- Epoch = 5 -----
Val loss = 0.025957
New best epoch, loss = 0.025957
----- Epoch = 6 -----
Val loss = 0.017574
New best epoch, loss = 0.017574
----- Epoch = 7 -----
Val loss = 0.015273
New best epoch, loss = 0.015273
----- Epoch = 8 -----
Val loss = 0.014053
New best epoch, loss = 0.014053
----- Epoch = 9 -----
Val loss = 0.013188
New best epoch, loss = 0.013188
----- Epoch = 10 -----
Val loss = 0.012649
New best epoch, loss = 0.012649
----- Epoch = 11 -----
Val loss = 0.012275
New best epoch, loss = 0.012275
----- Epoch = 12 -----
Val loss = 0.012005
New best epoch, loss = 0.012005
----- Epoch = 13 -----
Val loss = 0.011791
New best epoch, loss = 0.011791
----- Epoch = 14 -----
Val loss = 0.011636
New best epoch, loss = 0.011636
----- Epoch = 15 -----
Val loss = 0.011542
New best epoch, loss = 0.011542
----- Epoch = 16 -----
Val loss = 0.011452
New best epoch, loss = 0.011452
----- Epoch = 17 -----
Val loss = 0.011388
New best epoch, loss = 0.011388
----- Epoch = 18 -----
Val loss = 0.011344
New best epoch, loss = 0.011344
----- Epoch = 19 -----
Val loss = 0.011303
New best epoch, loss = 0.011303
----- Epoch = 20 -----
Val loss = 0.011292
New best epoch, loss = 0.011292
----- Epoch = 21 -----
Val loss = 0.011278
New best epoch, loss = 0.011278
----- Epoch = 22 -----
Val loss = 0.011252
New best epoch, loss = 0.011252
----- Epoch = 23 -----
Val loss = 0.011260
----- Epoch = 24 -----
Val loss = 0.011253
----- Epoch = 25 -----
Val loss = 0.011250
New best epoch, loss = 0.011250
----- Epoch = 26 -----
Val loss = 0.011243
New best epoch, loss = 0.011243
----- Epoch = 27 -----
Val loss = 0.011241
New best epoch, loss = 0.011241
----- Epoch = 28 -----
Val loss = 0.011233
New best epoch, loss = 0.011233
----- Epoch = 29 -----
Val loss = 0.011236
----- Epoch = 30 -----
Val loss = 0.011229
New best epoch, loss = 0.011229
----- Epoch = 31 -----
Val loss = 0.011229
New best epoch, loss = 0.011229
----- Epoch = 32 -----
Val loss = 0.011239
----- Epoch = 33 -----
Val loss = 0.011232
----- Epoch = 34 -----
Val loss = 0.011167
New best epoch, loss = 0.011167
----- Epoch = 35 -----
Val loss = 0.011160
New best epoch, loss = 0.011160
----- Epoch = 36 -----
Val loss = 0.011163
----- Epoch = 37 -----
Val loss = 0.011170
----- Epoch = 38 -----
Val loss = 0.011168
----- Epoch = 39 -----
Val loss = 0.011133
New best epoch, loss = 0.011133
----- Epoch = 40 -----
Val loss = 0.011129
New best epoch, loss = 0.011129
----- Epoch = 41 -----
Val loss = 0.011131
----- Epoch = 42 -----
Val loss = 0.011133
----- Epoch = 43 -----
Val loss = 0.011128
New best epoch, loss = 0.011128
----- Epoch = 44 -----
Val loss = 0.011136
----- Epoch = 45 -----
Val loss = 0.011136
----- Epoch = 46 -----
Val loss = 0.011137
----- Epoch = 47 -----
Val loss = 0.011119
New best epoch, loss = 0.011119
----- Epoch = 48 -----
Val loss = 0.011114
New best epoch, loss = 0.011114
----- Epoch = 49 -----
Val loss = 0.011113
New best epoch, loss = 0.011113
----- Epoch = 50 -----
Val loss = 0.011115
----- Epoch = 51 -----
Val loss = 0.011115
----- Epoch = 52 -----
Val loss = 0.011108
New best epoch, loss = 0.011108
----- Epoch = 53 -----
Val loss = 0.011101
New best epoch, loss = 0.011101
----- Epoch = 54 -----
Val loss = 0.011103
----- Epoch = 55 -----
Val loss = 0.011102
----- Epoch = 56 -----
Val loss = 0.011101
New best epoch, loss = 0.011101
----- Epoch = 57 -----
Val loss = 0.011100
New best epoch, loss = 0.011100
----- Epoch = 58 -----
Val loss = 0.011099
New best epoch, loss = 0.011099
----- Epoch = 59 -----
Val loss = 0.011098
New best epoch, loss = 0.011098
----- Epoch = 60 -----
Val loss = 0.011100
----- Epoch = 61 -----
Val loss = 0.011100
----- Epoch = 62 -----
Val loss = 0.011100
----- Epoch = 63 -----
Val loss = 0.011099
----- Epoch = 64 -----
Val loss = 0.011100
Stopping early at epoch = 63
We repeat the same figure plot like the previous case:
classes = adata.obs.str_labels.cat.categories
features = adata.var_names
attributions_class_pos_agg = pd.DataFrame()
n_cols = 3
top_n = 5
nrows = round(classes.size / n_cols)
fig, ax = plt.subplots(nrows, n_cols, sharex=False, figsize=(20, 20))
for idx, ct in enumerate(classes):
sum_shap_per_class = [0] * num_features
for ind in np.where(adata.obs.str_labels == ct)[0].tolist():
sum_shap_per_class += fastshap.shap_values(np.array([adata.X[ind].toarray()]))[0][:, 0]
attributions_class = pd.DataFrame(
{"gene": features, "mean_shap": sum_shap_per_class / num_features, "class": ct}
).sort_values("mean_shap", ascending=False)
positive = attributions_class.head(top_n)
positive["contribution"] = "positive"
negative = attributions_class.tail(top_n)
negative["contribution"] = "negative"
avg = pd.concat([positive, negative])
title = f"IG importance for: {ct}"
# also keep the positive contributions
attributions_class_pos = attributions_class[attributions_class.mean_shap > 0]
attributions_class_pos["class"] = ct
attributions_class_pos_agg = pd.concat([attributions_class_pos_agg, attributions_class_pos])
sns.barplot(
x="mean_shap",
y="gene",
hue="contribution",
palette=["blue", "red"],
data=avg,
ax=ax[idx // n_cols, idx % n_cols],
)
ax[idx // n_cols, idx % n_cols].set_title(title)
ax[idx // n_cols, idx % n_cols].legend(title="SHAP Contribution", loc="lower right")
_ = [fig.delaxes(ax_) for ax_ in ax.flatten() if not ax_.has_data()]
fig.tight_layout()

top_n = 20
# Pivot the data so that each group becomes a column for stacking
pivot_df = attributions_class_pos_agg.pivot_table(
index="gene", columns="class", values="mean_shap", aggfunc="sum"
)
# Sort by the total sum of each feature (sum across all groups)
pivot_df["total"] = pivot_df.sum(axis=1) # Calculate the total sum for each feature
pivot_df = pivot_df.sort_values(by="total", ascending=False) # Sort by the total value
pivot_df = pivot_df.head(top_n) # Select the top 10 features
# Plotting the horizontal stacked bar plot
ax = pivot_df.drop("total", axis=1).plot(
kind="barh", stacked=True, figsize=(10, 6), colormap="tab20"
)
# Add labels and title
ax.set_xlabel("SHAP Contribution Value")
ax.set_ylabel("Gene")
ax.set_title("Top 10 Stacked SHAP Contributions by Cell Type per Gene")
# Display the plot
plt.tight_layout()
plt.show()

And we can see some overlapping genes from the 2 methods for this specific group of cells
As for SCVI-tools v1.3 Work on SHAP is still in progress: please check back in the next release!