MrVI analysis over Tahoe100M cells dataset using LaminDB Custom Dataloader#
MrVI (Multi-resolution Variational Inference) is a model for analyzing multi-sample single-cell RNA-seq data. This tutorial show how to do run MrVI in PyTorch version over the Tahoe100M cells dataset and perform basic analysis, using Lamin custom dataloader.
!pip install --quiet scvi-colab
from scvi_colab import install
install()
import gc
import tempfile
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import scvi
import scvi.hub
import seaborn as sns
import torch
from scvi.dataloaders import MappedCollectionDataModule
from scvi.external import MRVI
run_autotune = False
# import inspect
# print(inspect.getsource(MRVI))
# os.system("lamin init --storage ./lamindb_collection")
import lamindb as ln
# ln.setup.init()
→ connected lamindb: anonymous/lamindb_collection
scvi.settings.seed = 0
print("Last run with scvi-tools version:", scvi.__version__)
Last run with scvi-tools version: 1.3.3
sc.set_figure_params(figsize=(6, 6), frameon=False)
sns.set_theme()
torch.set_float32_matmul_precision("high")
save_dir = tempfile.TemporaryDirectory()
%config InlineBackend.print_figure_kwargs={"facecolor": "w"}
%config InlineBackend.figure_format="retina"
pd.set_option("display.max_rows", 50)
pd.set_option("display.max_columns", 50)
pd.set_option("display.width", 1000)
Get the data#
We start by downloading the model from its hub in order to use its metadata Note that the model is very large therefore it will take time to being download.
# get the hub data
tahoe_hubmodel = scvi.hub.HubModel.pull_from_huggingface_hub(
repo_name="vevotx/Tahoe-100M-SCVI-v1", cache_dir="."
)
tahoe_hubmodel.model.adata.obs.head()
INFO Loading model...
INFO File ./models--vevotx--Tahoe-100M-SCVI-v1/snapshots/b5283a73fbbed812a95264ace360da538b20af89/model.pt
already downloaded
| sample | species | gene_count | tscp_count | mread_count | bc1_wind | bc2_wind | bc3_wind | bc1_well | bc2_well | bc3_well | id | drugname_drugconc | drug | INT_ID | NUM.SNPS | NUM.READS | demuxlet_call | BEST.LLK | NEXT.LLK | DIFF.LLK.BEST.NEXT | BEST.POSTERIOR | SNG.POSTERIOR | SNG.BEST.LLK | SNG.NEXT.LLK | SNG.ONLY.POSTERIOR | DBL.BEST.LLK | DIFF.LLK.SNG.DBL | sublibrary | BARCODE | pcnt_mito | S_score | G2M_score | phase | pass_filter | dataset | _scvi_batch | _scvi_labels | _scvi_observed_lib_size | plate | Cell_Name_Vevo | Cell_ID_Cellosaur | observed_lib_size | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BARCODE_SUB_LIB_ID | |||||||||||||||||||||||||||||||||||||||||||
| 01_001_052-lib_1105 | smp_1783 | hg38 | 1878 | 2893 | 3284 | 1 | 1 | 52 | A1 | A1 | E4 | recgIHRi9MiCIr4CO | [('8-Hydroxyquinoline', 0.05, 'uM')] | 8-Hydroxyquinoline | 1.0 | 199.0 | 215.0 | singlet | -50.74 | -59.04 | 8.30 | -55.0 | 1.0 | -50.74 | -87.96 | 0.0 | -59.04 | 8.30 | lib_1105 | 01_001_052 | 0.019357 | 0.174603 | 0.179670 | G2M | full | 0 | 0 | 0 | 2893 | 4 | PANC-1 | CVCL_0480 | 2893 |
| 01_001_105-lib_1105 | smp_1783 | hg38 | 1765 | 2434 | 2764 | 1 | 1 | 105 | A1 | A1 | p2.A9 | recgIHRi9MiCIr4CO | [('8-Hydroxyquinoline', 0.05, 'uM')] | 8-Hydroxyquinoline | 3.0 | 137.0 | 140.0 | singlet | -37.97 | -42.41 | 4.44 | -43.0 | 1.0 | -37.97 | -64.52 | 0.0 | -42.41 | 4.44 | lib_1105 | 01_001_105 | 0.029581 | 0.297619 | 0.342857 | G2M | full | 0 | 0 | 0 | 2434 | 4 | SW480 | CVCL_0546 | 2434 |
| 01_001_165-lib_1105 | smp_1783 | hg38 | 3174 | 5691 | 6454 | 1 | 1 | 165 | A1 | A1 | p2.F9 | recgIHRi9MiCIr4CO | [('8-Hydroxyquinoline', 0.05, 'uM')] | 8-Hydroxyquinoline | 4.0 | 379.0 | 396.0 | singlet | -129.66 | -130.65 | 0.99 | -130.0 | 1.0 | -129.66 | -186.89 | 0.0 | -130.65 | 0.99 | lib_1105 | 01_001_165 | 0.031629 | 0.031746 | 0.099084 | G2M | full | 0 | 0 | 0 | 5691 | 4 | SW1417 | CVCL_1717 | 5691 |
| 01_003_094-lib_1105 | smp_1783 | hg38 | 1380 | 1804 | 2050 | 1 | 3 | 94 | A1 | A3 | H10 | recgIHRi9MiCIr4CO | [('8-Hydroxyquinoline', 0.05, 'uM')] | 8-Hydroxyquinoline | 7.0 | 122.0 | 125.0 | singlet | -31.79 | -33.98 | 2.19 | -36.0 | 1.0 | -31.79 | -49.36 | 0.0 | -33.98 | 2.19 | lib_1105 | 01_003_094 | 0.017738 | -0.063492 | 0.019780 | G2M | full | 0 | 0 | 0 | 1804 | 4 | SW1417 | CVCL_1717 | 1804 |
| 01_003_164-lib_1105 | smp_1783 | hg38 | 1179 | 1514 | 1715 | 1 | 3 | 164 | A1 | A3 | p2.F8 | recgIHRi9MiCIr4CO | [('8-Hydroxyquinoline', 0.05, 'uM')] | 8-Hydroxyquinoline | 8.0 | 87.0 | 93.0 | singlet | -28.99 | -27.07 | -1.92 | -34.0 | 1.0 | -28.99 | -41.61 | 0.0 | -27.07 | -1.92 | lib_1105 | 01_003_164 | 0.023118 | -0.075397 | -0.070879 | G1 | full | 0 | 0 | 0 | 1514 | 4 | A498 | CVCL_1056 | 1514 |
# Load Cell Line Metadata
cell_lines = pd.read_csv(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/cell_line_metadata.h5ad"
)
cell_lines.head()
| Unnamed: 0 | cell_name | Cell_ID_DepMap | Cell_ID_Cellosaur | Organ | Driver_Gene_Symbol | Driver_VarZyg | Driver_VarType | Driver_ProtEffect_or_CdnaEffect | Driver_Mech_InferDM | Driver_GeneType_DM | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | A549 | ACH-000681 | CVCL_0023 | Lung | CDKN2A | Hom | Deletion | DEL | LoF | Suppressor |
| 1 | 1 | A549 | ACH-000681 | CVCL_0023 | Lung | CDKN2B | Hom | Deletion | DEL | LoF | Suppressor |
| 2 | 2 | A549 | ACH-000681 | CVCL_0023 | Lung | KRAS | Hom | Missense | p.G12S | GoF | Oncogene |
| 3 | 3 | A549 | ACH-000681 | CVCL_0023 | Lung | SMARCA4 | Hom | Frameshift | p.Q729fs | LoF | Suppressor |
| 4 | 4 | A549 | ACH-000681 | CVCL_0023 | Lung | STK11 | Hom | Stopgain | p.Q37* | LoF | Suppressor |
In the next part we are creating artifacts from a subset of 1M cells per plate (and 5 plates) from the dataset and unite them to a collection. Artifacts and collections are the ways lamindb interactes with the data. From this point forward we will not use adatas again. The tutorial assumed those files were already ready (see for how to here)
# Init Lamin instance
ln.track()
→ loaded Transform('GMYJf4FqDHnZ0000'), re-started Run('5FGprgCv...') at 2025-08-20 15:14:06 UTC
• recommendation: to identify the notebook across renames, pass the uid: ln.track("GMYJf4FqDHnZ")
# We make a collection of artifactos from files that are stored on disk
artifact1 = ln.Artifact.from_anndata(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/tahoe100m_sample_1000000_plate1.h5ad",
key="part_1.h5ad",
).save()
artifact2 = ln.Artifact.from_anndata(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/tahoe100m_sample_1000000_plate2.h5ad",
key="part_2.h5ad",
).save()
artifact3 = ln.Artifact.from_anndata(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/tahoe100m_sample_1000000_plate3.h5ad",
key="part_3.h5ad",
).save()
artifact4 = ln.Artifact.from_anndata(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/tahoe100m_sample_1000000_plate4.h5ad",
key="part_4.h5ad",
).save()
artifact5 = ln.Artifact.from_anndata(
"/home/access/PycharmProjects/scvi-tools/Tahoe100M/tahoe100m_sample_1000000_rand.h5ad",
key="part_5.h5ad",
).save()
! calling anonymously, will miss private instances
→ returning existing artifact with same hash: Artifact(uid='sBdR09Afz0MqbLu50002', is_latest=True, key='part_1.h5ad', suffix='.h5ad', kind='dataset', otype='AnnData', size=17933581240, hash='fmr6LyB4ySH0Q7vA_zCZMy', n_observations=1000000, branch_id=1, space_id=1, storage_id=1, run_id=2, created_by_id=1, created_at=2025-08-07 13:24:38 UTC); to track this artifact as an input, use: ln.Artifact.get()
→ returning existing artifact with same hash: Artifact(uid='6PuwYTVC0rc8ik9h0002', is_latest=True, key='part_2.h5ad', suffix='.h5ad', kind='dataset', otype='AnnData', size=19205241784, hash='Kd0DjdW_GFI2QXzeaFePJ2', n_observations=1000000, branch_id=1, space_id=1, storage_id=1, run_id=2, created_by_id=1, created_at=2025-08-07 13:24:49 UTC); to track this artifact as an input, use: ln.Artifact.get()
→ returning existing artifact with same hash: Artifact(uid='Y4Ht6CkaiGsXWWTJ0000', is_latest=True, key='part_3.h5ad', suffix='.h5ad', kind='dataset', otype='AnnData', size=15146243320, hash='f3_p_AkcSfh-P2aKlmOkeC', n_observations=1000000, branch_id=1, space_id=1, storage_id=1, run_id=2, created_by_id=1, created_at=2025-08-07 13:25:00 UTC); to track this artifact as an input, use: ln.Artifact.get()
→ returning existing artifact with same hash: Artifact(uid='GBKhWVRoRltw7UZk0001', is_latest=True, key='part_4.h5ad', suffix='.h5ad', kind='dataset', otype='AnnData', size=15207127144, hash='rJLJdKpkSvkvB-ks7A2z3e', n_observations=1000000, branch_id=1, space_id=1, storage_id=1, run_id=2, created_by_id=1, created_at=2025-08-07 13:25:11 UTC); to track this artifact as an input, use: ln.Artifact.get()
→ returning existing artifact with same hash: Artifact(uid='NBFiWx43U3hNm4dR0001', is_latest=True, key='part_5.h5ad', suffix='.h5ad', kind='dataset', otype='AnnData', size=17240836184, hash='7RX84cxLdX92Sb9Cu74ZjF', n_observations=1000000, branch_id=1, space_id=1, storage_id=1, run_id=2, created_by_id=1, created_at=2025-08-07 13:25:20 UTC); to track this artifact as an input, use: ln.Artifact.get()
collection = ln.Collection([artifact1, artifact2, artifact3, artifact4, artifact5], key="gather")
collection.save()
! returning existing collection with same hash: Collection(uid='6IASvDXmFbji3Uzu000Q', is_latest=True, key='gather', hash='5K17S6WOKo9udA6RiGW4Lw', branch_id=1, space_id=1, created_by_id=1, run_id=2, created_at=2025-08-07 13:25:33 UTC); if you intended to query to track this collection as an input, use: ln.Collection.get()
Collection(uid='6IASvDXmFbji3Uzu000Q', is_latest=True, key='gather', hash='5K17S6WOKo9udA6RiGW4Lw', branch_id=1, space_id=1, created_by_id=1, run_id=2, created_at=2025-08-07 13:25:33 UTC)
# We load the collection to see it consists of many h5ad files
artifacts = collection.artifacts.all()
artifacts.df()
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | _hash_type | _key_is_virtual | _overwrite_versions | space_id | storage_id | schema_id | version | is_latest | run_id | created_at | created_by_id | _aux | branch_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||||
| 39 | sBdR09Afz0MqbLu50002 | part_1.h5ad | None | .h5ad | dataset | AnnData | 17933581240 | fmr6LyB4ySH0Q7vA_zCZMy | None | 1000000 | sha1-fl | True | False | 1 | 1 | None | None | True | 2 | 2025-08-07 13:24:38.913000+00:00 | 1 | None | 1 |
| 40 | 6PuwYTVC0rc8ik9h0002 | part_2.h5ad | None | .h5ad | dataset | AnnData | 19205241784 | Kd0DjdW_GFI2QXzeaFePJ2 | None | 1000000 | sha1-fl | True | False | 1 | 1 | None | None | True | 2 | 2025-08-07 13:24:49.589000+00:00 | 1 | None | 1 |
| 41 | Y4Ht6CkaiGsXWWTJ0000 | part_3.h5ad | None | .h5ad | dataset | AnnData | 15146243320 | f3_p_AkcSfh-P2aKlmOkeC | None | 1000000 | sha1-fl | True | False | 1 | 1 | None | None | True | 2 | 2025-08-07 13:25:00.960000+00:00 | 1 | None | 1 |
| 42 | GBKhWVRoRltw7UZk0001 | part_4.h5ad | None | .h5ad | dataset | AnnData | 15207127144 | rJLJdKpkSvkvB-ks7A2z3e | None | 1000000 | sha1-fl | True | False | 1 | 1 | None | None | True | 2 | 2025-08-07 13:25:11.405000+00:00 | 1 | None | 1 |
| 43 | NBFiWx43U3hNm4dR0001 | part_5.h5ad | None | .h5ad | dataset | AnnData | 17240836184 | 7RX84cxLdX92Sb9Cu74ZjF | None | 1000000 | sha1-fl | True | False | 1 | 1 | None | None | True | 2 | 2025-08-07 13:25:20.940000+00:00 | 1 | None | 1 |
we can now define the batch and data loader which replaces the default AnnDataloder of and use that on MRVI model.
datamodule = MappedCollectionDataModule(
collection,
batch_key="plate",
sample_key="sample",
batch_size=1024,
shuffle=True,
join="inner",
model_name="TorchMRVI",
collection_val=collection,
)
print(datamodule.n_obs, datamodule.n_vars, datamodule.n_batch)
5000000 62710 14
Train mrVI with LaminDB#
We will initialize the MRVI model with its “pytorch” backend. A JAX backend version can be also be used using backend=”jax”.
# Init the model
model = MRVI(registry=datamodule.registry, backend="torch")
# Training the model (for 5M cells will take 1 day+ with early stopping - better to cancel it)
import time
gc.collect()
start = time.time()
model.train(
max_epochs=50,
# early_stopping=True,
plan_kwargs={"lr": 1e-3, "n_epochs_kl_warmup": 40},
datamodule=datamodule,
batch_size=1024,
# early_stopping_patience=5,
# check_val_every_n_epoch=1,
# datasplitter_kwargs={
# "external_indexing": [np.array(train_ind), np.array(valid_ind)]
# }
)
end = time.time()
print(f"Elapsed time: {end - start:.2f} seconds")
Elapsed time: 75375.09 seconds
model.history.keys()
dict_keys(['kl_weight', 'train_loss_step', 'validation_loss', 'elbo_validation', 'reconstruction_loss_validation', 'kl_local_validation', 'kl_global_validation', 'train_loss_epoch', 'elbo_train', 'reconstruction_loss_train', 'kl_local_train', 'kl_global_train'])
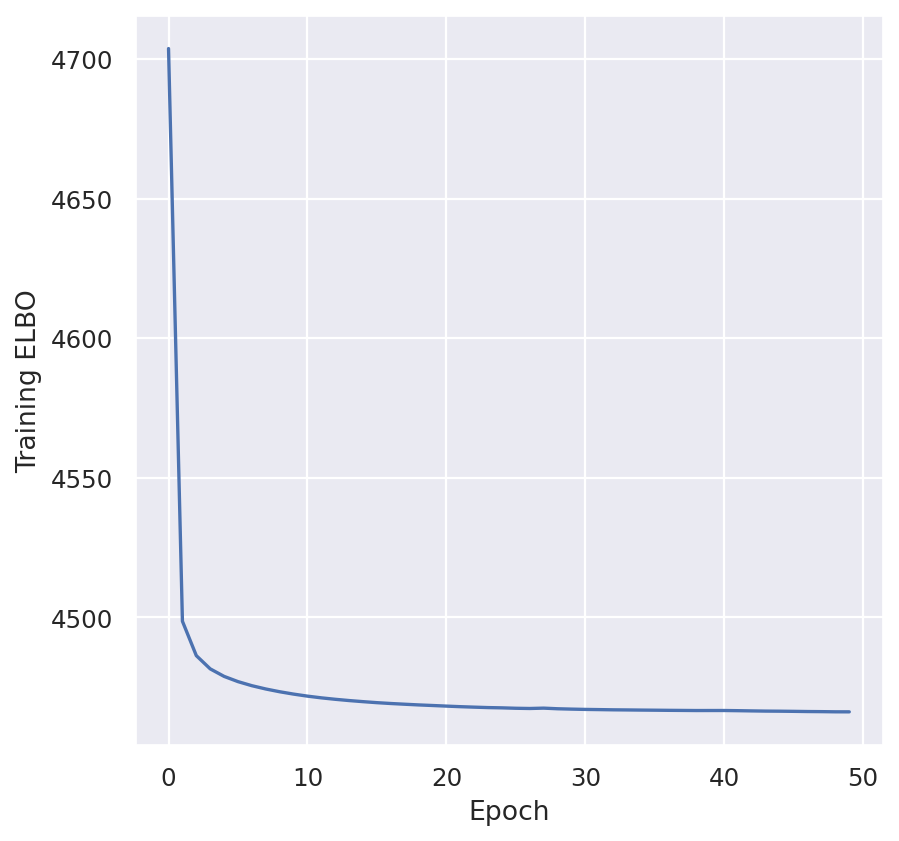
plt.plot(model.history["elbo_validation"])
plt.xlabel("Epoch")
plt.ylabel("Validation ELBO")
plt.show()
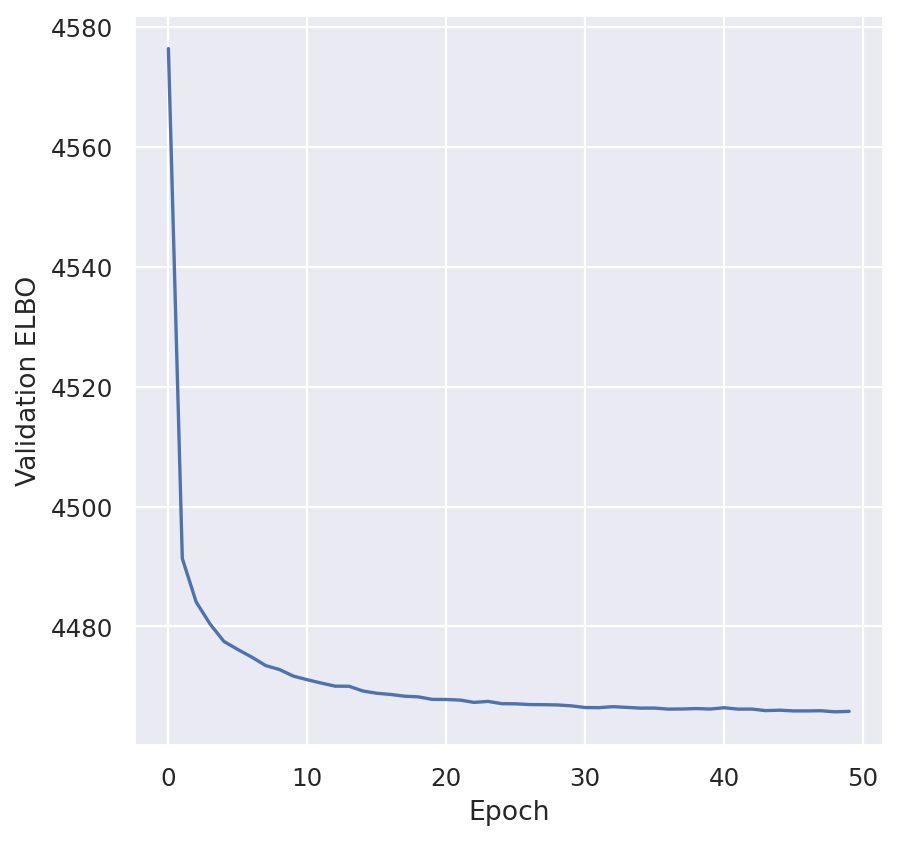
plt.plot(model.history["reconstruction_loss_validation"])
plt.xlabel("Epoch")
plt.ylabel("Validation reconstruction_loss")
plt.show()
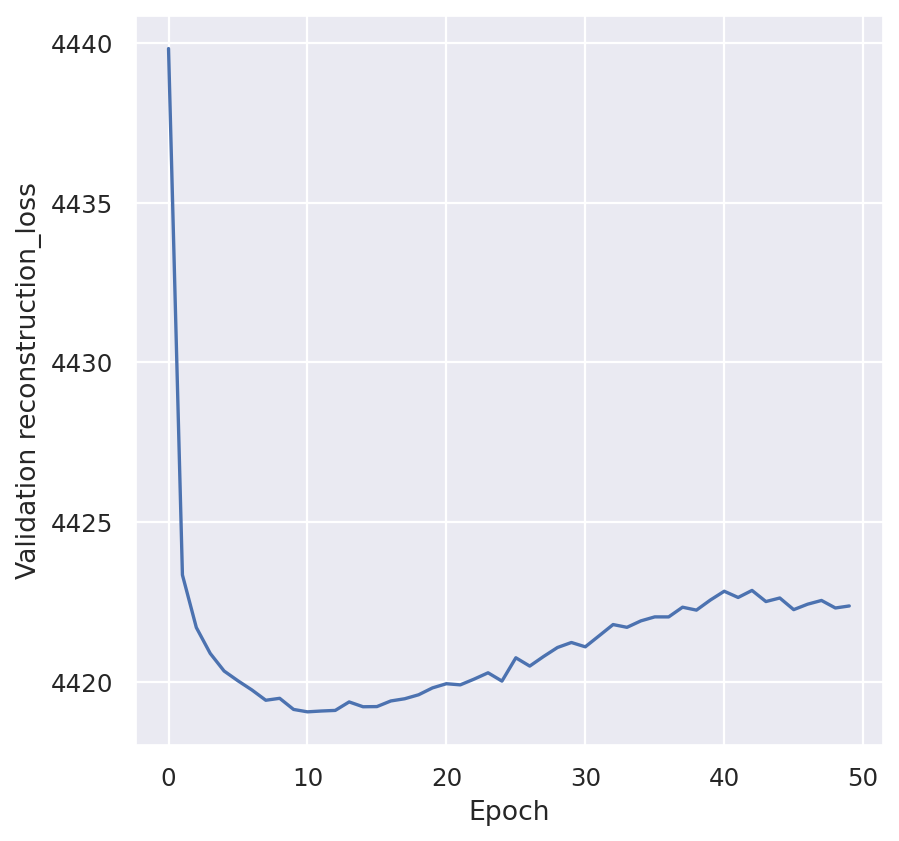
plt.plot(model.history["kl_local_validation"])
plt.xlabel("Epoch")
plt.ylabel("Validation KL")
plt.show()
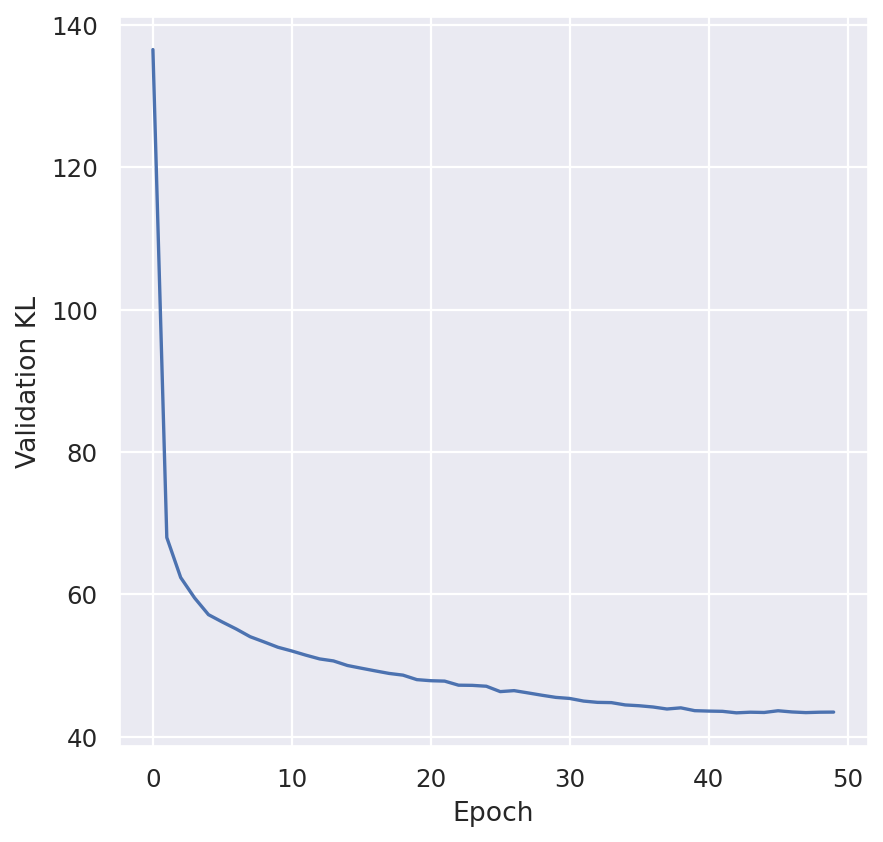
plt.plot(model.history["elbo_train"])
plt.xlabel("Epoch")
plt.ylabel("Training ELBO")
plt.show()
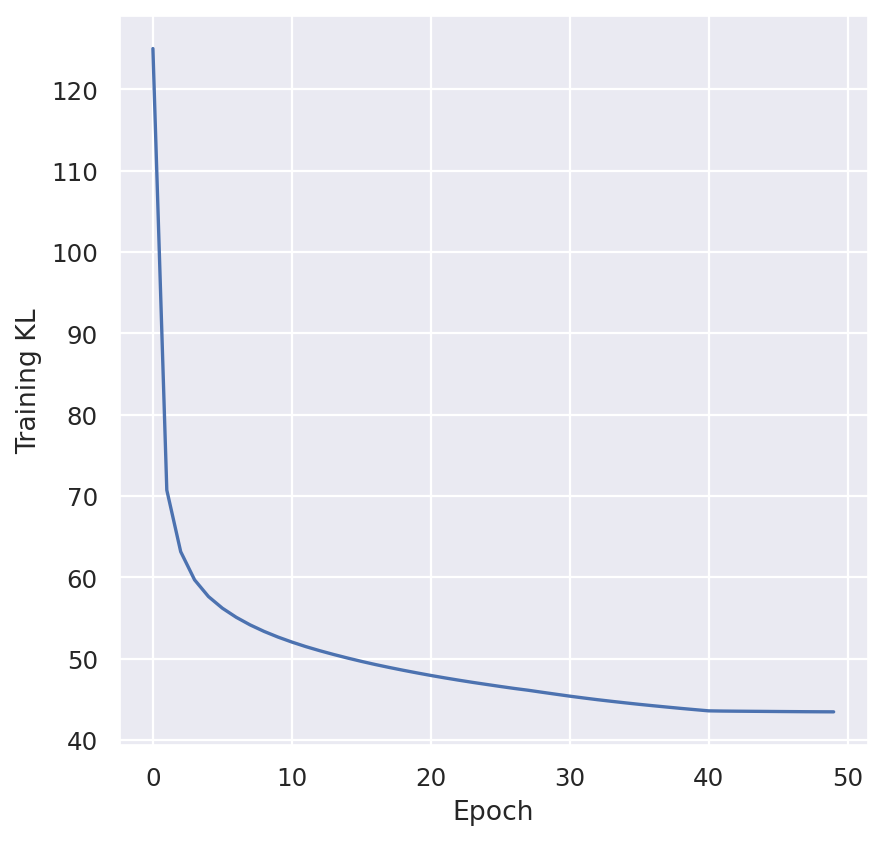
plt.plot(model.history["kl_local_train"])
plt.xlabel("Epoch")
plt.ylabel("Training KL")
plt.show()
# Save the model
model.save(
"mrvi_torch_tahoe100_lamin_model", save_anndata=False, overwrite=True, datamodule=datamodule
)
# Load the model
# model = MRVI.load("mrvi_torch_tahoe100_lamin_model", adata=False)
# We extract the adata of the model, to be able to use it for plot umaps
# To save time we could also select a sub set of it
adata = collection.load(join="inner")
adata
AnnData object with n_obs × n_vars = 5000000 × 62710
obs: 'drug', 'sample', 'BARCODE_SUB_LIB_ID', 'cell_line_id', 'moa-fine', 'canonical_smiles', 'pubchem_cid', 'plate', 'mean_gene_count', 'mean_tscp_count', 'mean_mread_count', 'mean_pcnt_mito', 'drugname_drugconc', 'targets', 'moa-broad', 'human-approved', 'clinical-trials', 'gpt-notes-approval', 'artifact_uid'
adata.obs.plate.value_counts()
plate
plate4 1141125
plate2 1084672
plate3 1056448
plate1 1028224
plate5 125045
plate10 112896
plate12 112896
plate11 84672
plate9 56450
plate7 56450
plate14 56448
plate6 28225
plate8 28225
plate13 28224
Name: count, dtype: int64
# merge metadata (will add memory)
# adata.obs = adata.obs.merge(tahoe_hubmodel.model.adata.obs[["Cell_Name_Vevo",
# "dataset","phase","observed_lib_size","S_score","G2M_score","sublibrary"]],
# how='left', left_on='BARCODE_SUB_LIB_ID', right_index=True)
adata
AnnData object with n_obs × n_vars = 5000000 × 62710
obs: 'drug', 'sample', 'BARCODE_SUB_LIB_ID', 'cell_line_id', 'moa-fine', 'canonical_smiles', 'pubchem_cid', 'plate', 'mean_gene_count', 'mean_tscp_count', 'mean_mread_count', 'mean_pcnt_mito', 'drugname_drugconc', 'targets', 'moa-broad', 'human-approved', 'clinical-trials', 'gpt-notes-approval', 'artifact_uid'
# In order to save memory for the sake of this tutorial we drop the
# count matrix from this adata (like done during minification)
from scipy.sparse import csr_matrix
del adata.raw
adata.X = csr_matrix(adata.X.shape)
# The way to extract the internal model analysis is by the inference_dataloader
# Datamodule will always require to pass it into all downstream functions.
inference_dataloader = datamodule.inference_dataloader(
batch_size=1024, parallel_cpu_count=5, shuffle=False
)
gc.collect()
9430
latent_representation = model.get_latent_representation(
give_z=False, dataloader=inference_dataloader
)
latent_representation.shape
(5000000, 10)
We removed the count layer from the adata therefore we cant run PCA like before
# adata.layers["counts"] = adata.X.copy() # preserve counts
# sc.pp.normalize_total(adata, target_sum=1e4)
# sc.pp.log1p(adata)
# adata.raw = adata # freeze the state in `.raw`
# run PCA then generate UMAP plots
# sc.tl.pca(adata)
# sc.pp.neighbors(adata)
# sc.tl.umap(adata, min_dist=0.1)
# sc.pl.umap(
# adata,
# color=["plate", "cell_line_id"],
# ncols=2,
# frameon=False,
# )
adata.obsm["X_mrVI_Torch_Lamin"] = latent_representation
# Subsample the adata to save time and memory
adata_subsampled = adata[
list(np.random.choice(np.arange(adata.n_obs), size=100000, replace=False)), :
].copy()
adata_subsampled.obsm["X_mrVI_Torch_Lamin"].shape
(100000, 10)
sc.pp.neighbors(adata_subsampled, use_rep="X_mrVI_Torch_Lamin")
sc.tl.umap(adata_subsampled, min_dist=0.3)
sc.pl.umap(
adata_subsampled,
color=["plate", "cell_line_id"],
frameon=False,
ncols=2,
)
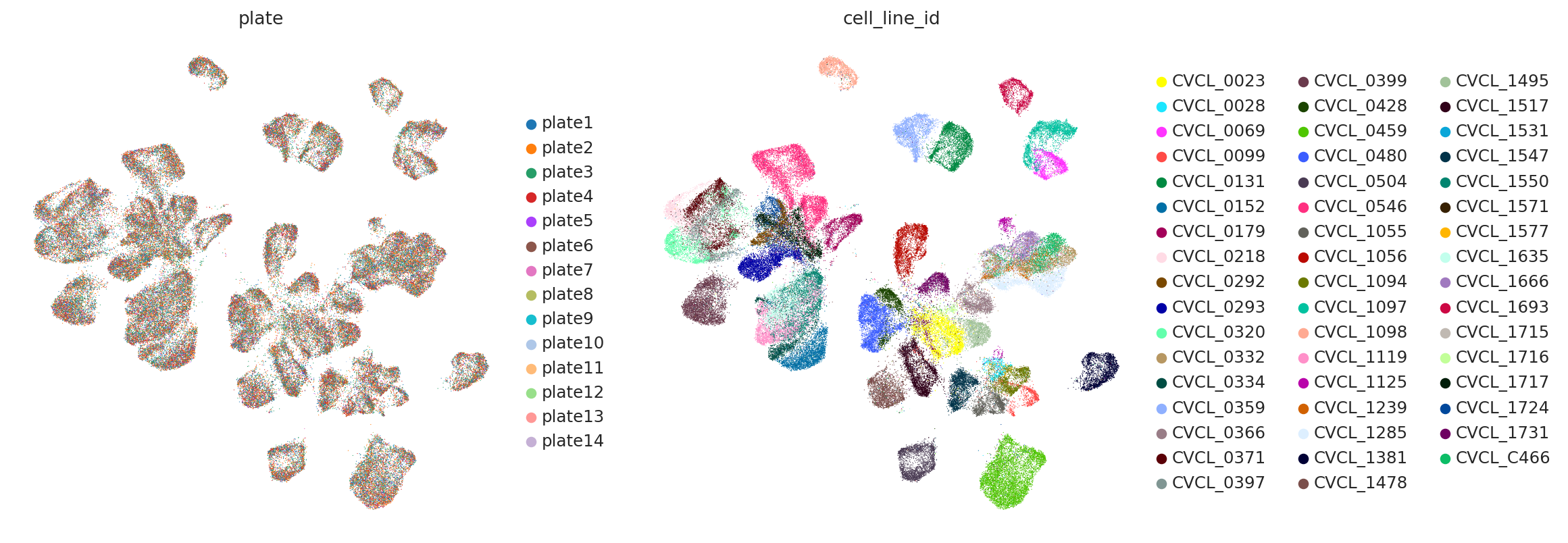
We also didnt use the metadata
# sc.pl.umap(
# adata,
# color=["moa-broad","phase"],
# frameon=False,
# ncols=2,
# )
# sc.pl.umap(
# adata,
# color=["observed_lib_size","S_score","G2M_score"],
# frameon=False,
# ncols=3,
# )
Compare results#
from scib_metrics.benchmark import BatchCorrection, Benchmarker, BioConservation
bm = Benchmarker(
adata[list(np.random.choice(np.arange(adata.n_obs), size=10000, replace=False)), :],
batch_key="plate",
bio_conservation_metrics=BioConservation(
isolated_labels=True,
nmi_ari_cluster_labels_leiden=True,
silhouette_label=True,
clisi_knn=True,
nmi_ari_cluster_labels_kmeans=True,
),
batch_correction_metrics=BatchCorrection(
bras=True,
pcr_comparison=True,
kbet_per_label=True,
graph_connectivity=False,
ilisi_knn=True,
),
label_key="cell_line_id",
embedding_obsm_keys=["X_mrVI_Torch_Lamin"],
n_jobs=-1,
)
bm.benchmark()
bm.plot_results_table(min_max_scale=False)